Biomarkers for Senescent Cell Burden
Senescent cells matter because they are not merely old cells sitting quietly at the edge of relevance. They alter local signaling, reshape immune traffic, and push nearby tissue toward inflammation, fibrosis, or repair failure. The difficulty is measurement. No single biomarker identifies all senescent cells across all tissues, and no clean blood test yet captures total burden with clinical precision.
That creates a familiar problem in longevity medicine. A mechanism can be biologically important while its measurement stack remains immature. Senescence is now in that phase. The field knows enough to reject the view that burden is unknowable. It still does not have a universal assay that turns senescence into one decision-grade number.
Established fact: senescent cells are heterogeneous across tissue, trigger, and secretory profile, and the standard markers such as p16, p21, and SA-beta-gal each capture only part of the state. Reasoned inference: the most useful measurement systems will be layered panels that combine cell-state markers, secreted factors, tissue context, and change over time rather than one canonical test.
The Core Measurement Problem
A useful biomarker has to do more than correlate loosely with age. It has to tell the reader something operational. Does burden appear elevated in a specific tissue? Does the burden look inflammatory, fibrotic, or immunologically trapped? Does the signal move after intervention in a way that lines up with function rather than with assay noise alone?
Single markers fail because senescence is not one molecular identity. Cells can be growth-arrested for different reasons. They can express DNA-damage programs, lysosomal expansion, secretory factors, or checkpoint proteins in different combinations. Even within one tissue, a senescent endothelial cell, adipocyte progenitor, fibroblast, or immune cell can look biologically related while still presenting a different assay profile.
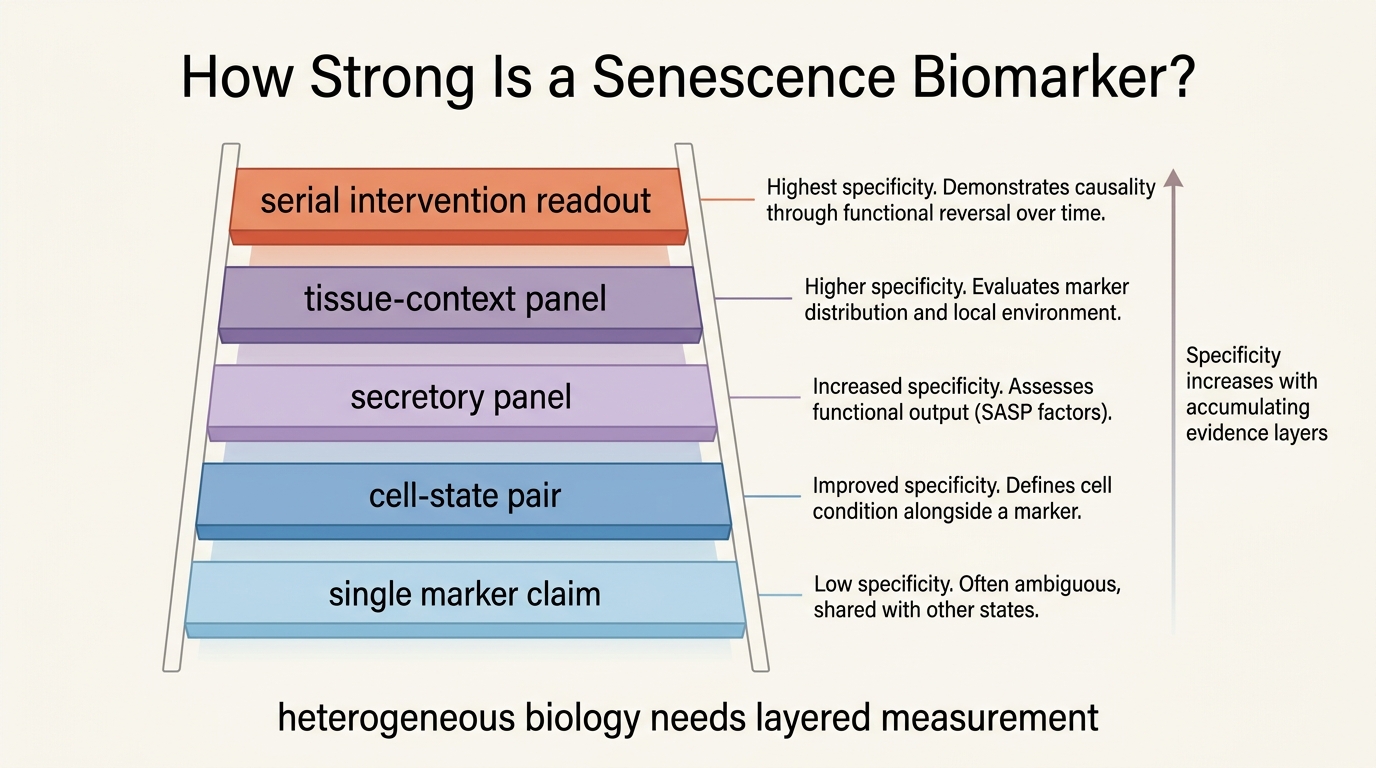
What the Main Marker Classes Actually Tell You
The checkpoint markers are the most familiar starting point. p16INK4a and p21 help identify durable cell-cycle arrest, and they matter because senescence is partly defined by that arrest logic. They are still insufficient on their own. Some senescent cells do not show strong expression at the same moment or in the same tissue, and some stressed non-senescent cells can transiently overlap with parts of the pattern.
Lysosomal markers such as SA-beta-gal add another layer because senescent cells often show characteristic lysosomal expansion. That is useful in tissue work and microscopy. It is weaker as a universal summary because lysosomal activity is not unique to senescence, and assay quality varies with sample handling and cell type.
Then there is the secretory layer, commonly grouped under the senescence-associated secretory phenotype. Cytokines, chemokines, matrix-remodeling proteins, and growth factors matter because they are often what make senescence harmful at tissue scale. The problem is that SASP factors are context-loaded. Inflammation, obesity, infection, cancer therapy, and wound repair can all move related markers without implying identical senescent burden.
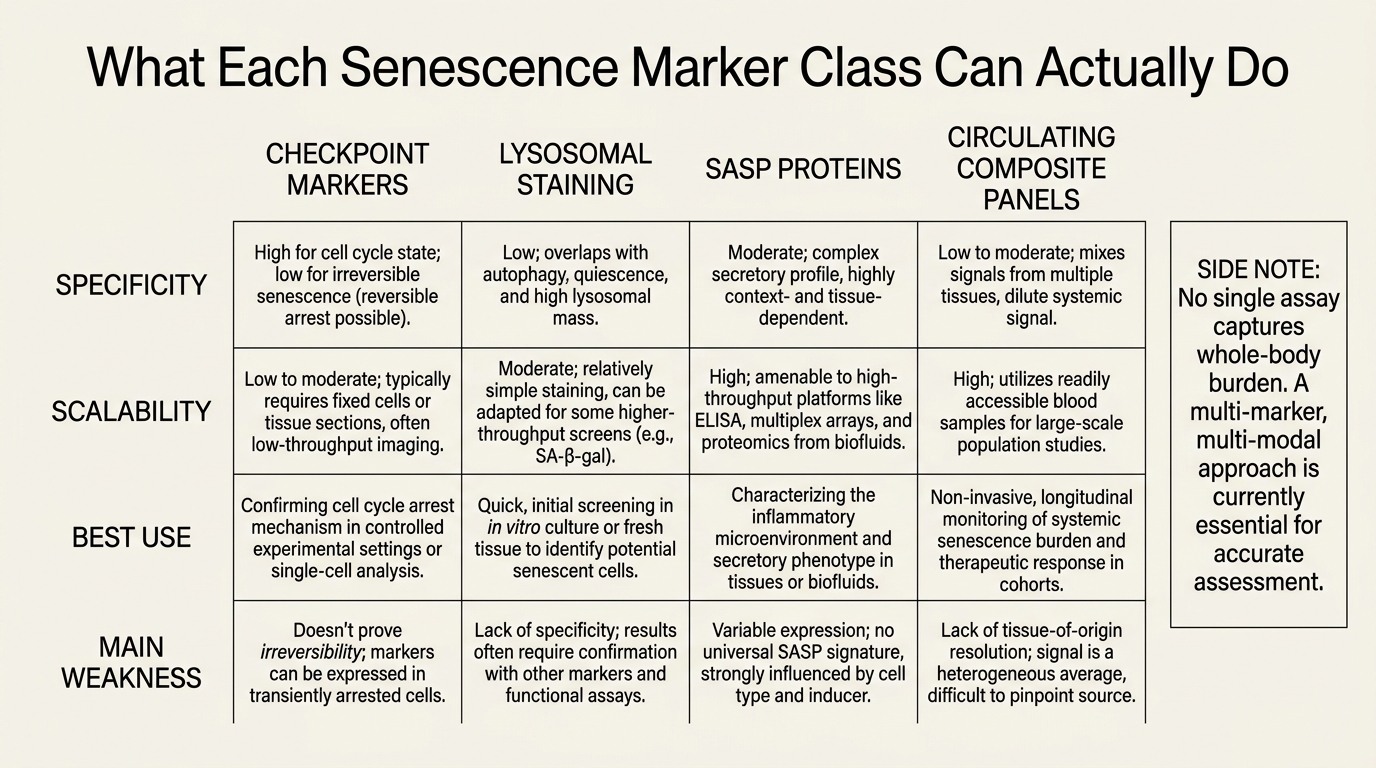
| Marker class | What it captures best | Main weakness |
|---|---|---|
| p16, p21, checkpoint expression | Durable growth arrest and senescence program engagement | Not universal across tissues or moments in the same tissue |
| SA-beta-gal and lysosomal staining | Classical cellular senescence phenotype in tissue or cell assays | Good microscopy signal does not equal whole-body burden measurement |
| SASP proteins and inflammatory panels | How senescence may be exerting downstream damage | Inflammation is not specific to senescence |
| DNA-damage, telomere, chromatin features | Upstream stress architecture around the senescent state | Stress signatures can persist outside true senescence |
| Circulating composite panels | Practical serial monitoring across time | Still too indirect to claim exact body-wide burden |
Why Panels Will Beat Single Markers
The best current path is not choosing one winner. It is combining orthogonal evidence. A stronger senescence panel might pair checkpoint expression, secretory pattern, tissue-specific context, and functional correlates such as fibrosis, endothelial dysfunction, or impaired repair. That approach does not solve every specificity problem, but it lowers the chance of mistaking generic stress for true senescent burden.
This is also where blood-based monitoring becomes more realistic. Blood panels can still matter, especially if the goal is trend detection before and after intervention. They just need to be interpreted as partial windows into a distributed tissue process. A plasma score that moves after therapy is informative. It is not the same thing as counting every senescent cell that exists in the body.
What Would Make a Biomarker Clinically Useful
A clinically useful senescence biomarker has to clear a narrower bar than many longevity readers assume. It does not need to solve the philosophy of aging. It needs to predict something actionable. A good assay should help stratify risk, support intervention selection, or verify that a therapy changed the biological layer it claimed to target.
That means three validation questions matter more than branding. First, does the marker reproduce reliably within the same person? Second, does it line up with meaningful tissue or disease outcomes? Third, does it respond to intervention in a way that exceeds generic inflammatory fluctuation? Many current claims pass one question and fail the others.
For that reason, readers should be skeptical when a clinic or supplement company markets a senescence score as if the field already has a blood-pressure equivalent for cellular aging. It does not. The real progress is narrower and still significant: panels are getting better, tissue logic is getting clearer, and the gap between preclinical senescence biology and human measurement is shrinking.
How to Read Senolytic Claims
This measurement problem feeds directly into the senolytic debate. If the assay is weak, a therapy can look more confident than it really is. A small improvement in an inflammatory marker does not prove broad senescent-cell clearance. A biopsy signal in one compartment does not prove organism-wide rejuvenation. The right standard is convergence. Look for multiple marker layers, tissue relevance, and functional follow-through.
That is why the most serious senescence work still looks careful rather than triumphant. It treats burden as a contextual systems variable. In practice that means pairing senescence biology with tissue-specific interpretation, exactly the move developed in Tissue-Specific Senescence: Why Location Matters and in the translational reality check inside Senolytics Moving into Clinical Translation.
What Is Known, What Is Inferred, What Is Missing
What is known is that senescent burden is biologically real, heterogeneous, and consequential. What is inferred is that decision-grade monitoring will likely come from multi-marker panels combined with tissue logic rather than from one universal assay. What remains missing is a scalable, validated framework that lets a clinician measure senescent burden, intervene, and verify outcome with the same confidence now expected in mature specialties.
That is still progress. The field is moving from metaphor toward instrumentation. It is just not yet at the point where one number can summarize the burden honestly.
Source Frame
Established claims in this article draw on the mature senescence literature around p16 and p21 checkpoint signaling, SA-beta-gal staining, SASP heterogeneity, and the translational limits of blood-only inference.
Interpretive claims in this article are reasoned from that literature: namely that clinical usefulness will come from layered panels, repeated measures, and tissue context rather than from one universal biomarker.
Translate this longevity claim into a capital-runway decision.
Life extension logic only matters if the balance sheet can carry it. Move into WealthMeter to compare assets, spending, and yield assumptions against the same long-horizon planning problem.