Epigenetic Reprogramming Enters Human Trials
Rejuvenation research has crossed a threshold. The question is no longer whether partial reprogramming can reset age-associated biology in animals. It is now whether it can meet human safety and functional standards.
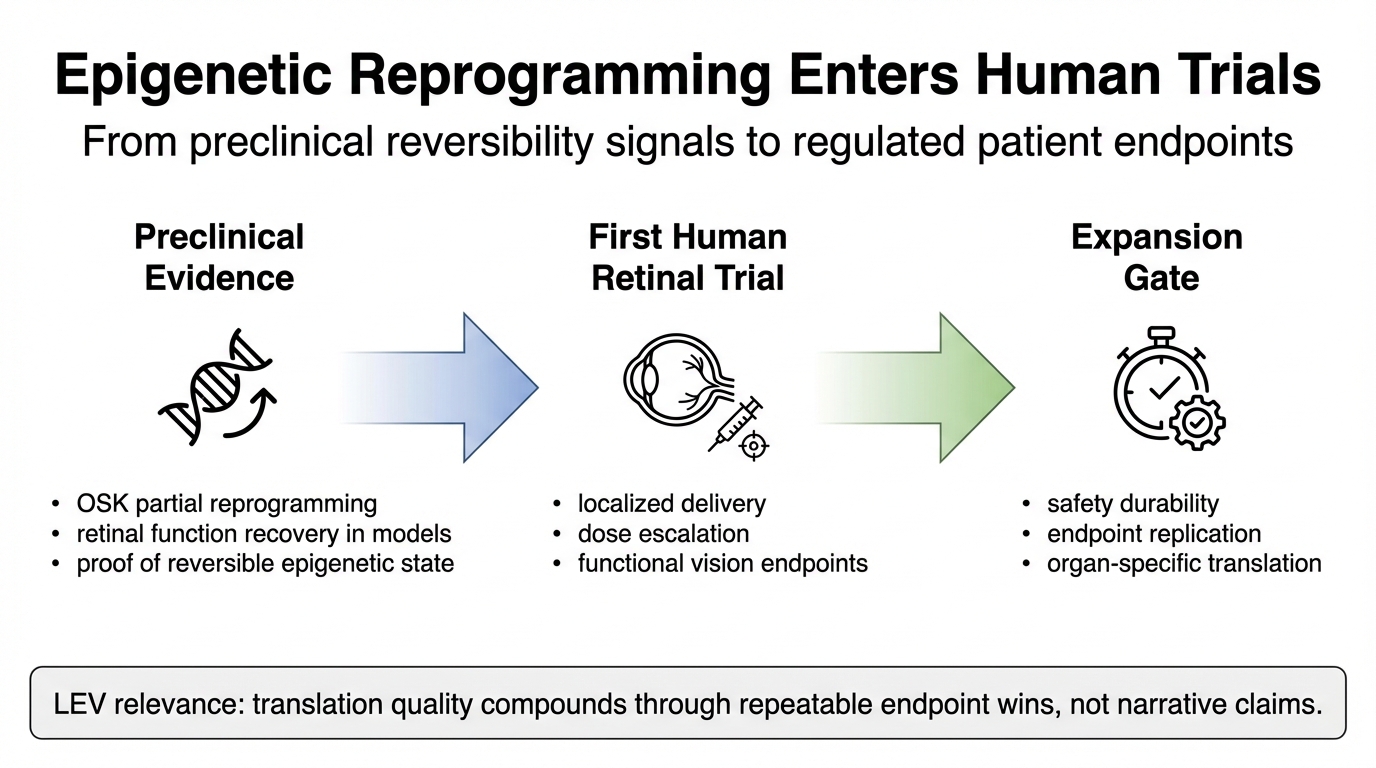
For more than a decade, epigenetic reprogramming has been the most promising concept in longevity science: if aging is partly encoded in reversible gene-expression states, then biological age may be modifiable in principle. That idea gained scientific traction after Nobel-recognized reprogramming work and later animal studies showing that partial reprogramming can improve tissue function without forcing full dedifferentiation. In January 2026, the field moved from theory and mice into human trials when Life Biosciences announced that the FDA cleared its first-in-human ER-100 trial for age-related eye disease.
What Is Established
Animal evidence is significant
Several preclinical studies support the claim that partial reprogramming can restore aspects of tissue function in specific contexts. Ocampo et al. reported that cyclic expression of Yamanaka factors improved multiple aging hallmarks and extended lifespan in a progeroid mouse model. Lu et al. reported vision recovery in mouse models of optic nerve injury and glaucoma using reprogramming logic focused on epigenetic information restoration. These results are not direct proof of whole-body human rejuvenation, but they are strong enough to justify tightly scoped human translation efforts. Additional studies in muscle, brain, and cardiovascular tissues have reinforced the potential for epigenetic modulation to reverse age-related functional decline, though with varying degrees of efficacy and reproducibility across tissue types.
Regulatory entry has started, but only in a narrow indication
Life Biosciences reported FDA clearance to begin ER-100 testing in retinal disease using OSK factors. This matters because retinal disease offers a practical first target: local delivery is more feasible than systemic exposure, imaging endpoints are rich, and functional testing is clinically mature. Regulators can evaluate safety and efficacy with greater precision than in systemic aging claims. The selection of ophthalmology as an entry point reflects a broader strategic pattern in gene therapy development, where localized administration reduces systemic risks and simplifies drug behavior modeling.
Clinical relevance now depends on endpoint quality
The human translation bar is not methylation changes alone. It is durable function under an acceptable risk profile. For eye indications, that means visual function, imaging-confirmed tissue behavior, adverse-event surveillance, and dose controls that reduce cancer and off-target risk. Any credible path to broader rejuvenation medicine will have to pass through this endpoint discipline repeatedly, organ by organ. The transition from biomarker-driven exploratory studies to clinically validated endpoints represents a maturation phase for the entire rejuvenation biotechnology sector, imposing rigorous standards on measurement precision and outcome durability.
What Is Inference, Not Yet Proof
It is reasonable to infer that success in retina could open adjacent indications with localized delivery and clear functional readouts. It is not yet reasonable to infer that systemic multi-organ rejuvenation is near clinical deployment. The biology, delivery constraints, and monitoring burden differ substantially across organs. The eye is a useful proving ground, not a universal solution. Extrapolation to systemic applications requires solving vector distribution, immunogenicity, and tissue-specific dosing challenges that remain largely unaddressed in current clinical frameworks. Moreover, epigenetic states vary by cell type, suggesting that reprogramming protocols may need significant customization for different tissues.
Evidence boundary: Current human evidence is early-stage and indication-specific. Claims of generalized age reversal in humans remain unproven.
Strategic implication: Track trial design quality and endpoint integrity rather than narrative intensity around the word “rejuvenation.”
Practical takeaway: Focus investment and attention on trials with robust functional endpoints and transparent data reporting, as these will set the evidentiary standard for the field.
Why This Milestone Is Different
Most longevity stories rely on theoretical projections. This one is practical. A first-in-human trial imposes regulatory requirements: manufacturing controls, protocol governance, adverse-event reporting, independent oversight, and long follow-up. Those requirements are exactly what convert laboratory promise into medical reality. If the field cannot perform under these constraints, the thesis remains scientific curiosity rather than healthcare infrastructure. The transition to regulated clinical development introduces accountability mechanisms that force precision in hypothesis testing and risk management, separating credible progress from speculative claims.
This is also where many public LEV narratives become testable. Longevity escape velocity is often described as compounding gains in healthy lifespan per calendar year. In practice, compounding requires repeated, validated clinical wins. A single dramatic preclinical result does not compound by itself. Reproducible safety engineering and endpoint discipline do. The ER-100 trial represents an initial step in a potential sequence of therapeutic approvals, each building on prior safety and efficacy data to enable more ambitious interventions.
Trial Design Realities That Will Decide Outcome
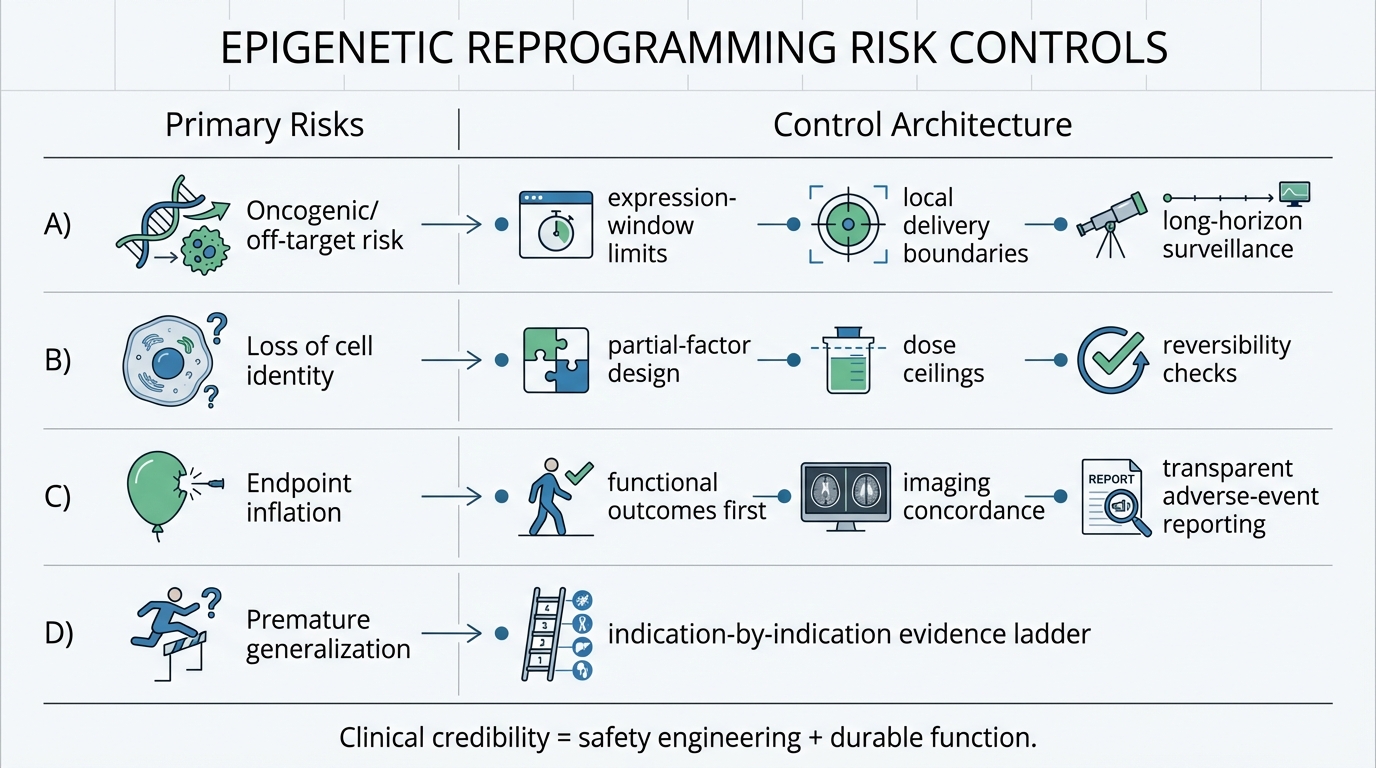
1) Dose and expression control
Partial reprogramming has to remain partial. The line between functional reset and loss of cell identity is a core risk boundary. Trial design has to enforce expression windows and exposure limits that preserve tissue identity while testing regenerative signal. This requires sophisticated delivery systems, such as controlled promoters or inducible vectors, to achieve transient and spatially controlled expression of reprogramming factors. Preclinical data suggest that even slight overexpression or prolonged exposure can lead to tumor formation or cellular dysfunction, underscoring the need for precision in clinical dosing regimens.
2) Functional outcomes over surrogate-only storytelling
Biomarkers can guide mechanism, but function determines patient value. Regulators, clinicians, and payers will weight durable visual outcomes and safety profiles more heavily than isolated molecular shifts. For retinal applications, this means measures like best-corrected visual acuity, contrast sensitivity, and reading speed, supported by structural improvements on optical coherence tomography. Relying solely on surrogate markers, such as DNA methylation clocks, without correlated functional benefits, is insufficient for regulatory approval or clinical adoption.
3) Long-horizon safety monitoring
Even when short-term tolerability is acceptable, age-modifying gene-therapy logic carries long-term uncertainty. Oncology, inflammation, and off-target surveillance will remain central for years after early readouts. The potential for delayed adverse events, such as neoplasia or autoimmune reactions, necessitates extended follow-up periods in trials and post-marketing studies. Real-world evidence frameworks will be critical for capturing rare or latency-period events that may not emerge during initial clinical testing.
What To Watch Over The Next 18 Months
Three signals are actionable and decision-relevant. First, trial execution quality: enrollment pace, protocol adherence, and transparent adverse-event reporting. Second, endpoint coherence: whether functional outcomes and biological measures move in a compatible direction. Third, replication trajectory: whether additional teams, indications, or platforms produce convergent evidence instead of isolated claims. Observers should also monitor regulatory interactions and guidance documents, as these will shape the development pathways for subsequent reprogramming-based therapies.
If these signals are positive, partial reprogramming moves from speculative frontier to a credible clinical platform class. If they are mixed, progress still continues but timeline assumptions for broad rejuvenation should be lengthened. Negative outcomes, such as significant safety issues or lack of efficacy, would necessitate a reevaluation of the reprogramming approach, though they might not invalidate the entire epigenetic reset concept if issues are addressable through improved delivery or factor selection.
LEV Placement: Compounding Phase, Not Endpoint Phase
Public forecasts for LEV timing vary widely. Ray Kurzweil has discussed late-2020s to mid-2030s windows in recent writing; Aubrey de Grey has also discussed non-trivial probabilities in the 2030s. Those forecasts are indicative. Clinical bottlenecks still determine realized timelines. The present phase looks like capability building in discovery and early translation, with access and validation as the key constraints. Each successful trial or approved therapy builds the foundational evidence and technical infrastructure needed for more complex multi-modal interventions, gradually accelerating the pace of progress.
The operational reading is straightforward: this is a meaningful milestone, but it is the beginning of medical validation, not the end of uncertainty. The transition from preclinical promise to clinical reality will be iterative, with each step requiring rigorous validation and often revealing new biological or logistical challenges. Stakeholders should expect a nonlinear path, with periods of rapid advancement followed by necessary consolidation and safety refinement.
Source List
Life Biosciences. (2026, January 13). FDA clearance announcement for ER-100 IND.
Ocampo, A., et al. (2016). In vivo amelioration of age-associated hallmarks by partial reprogramming. Cell.
Lu, Y., et al. (2020). Reprogramming to recover youthful epigenetic information and restore vision. Nature.
Nobel Prize. (2012). Press release: Physiology or Medicine 2012.
Nature Biotechnology. (2025). When does anti-ageing become medicine? (Perspective context on translational standards).