DNA Methylation Drift: Cause or Consequence of Aging?
DNA methylation drift is one of aging biology's most seductive signals because it looks both measurable and interpretable. Clocks built from methylation marks can predict chronological age unusually well. Reprogramming can reset many of those marks toward youth. Cancer and clonal expansion often carry adjacent methylation disturbance. It is tempting to collapse all of that into one strong conclusion: methylation drift causes aging. The evidence does not support that full claim.
The cleaner answer is layered. Some methylation change looks like accumulated history: replication, stress exposure, inflammation, mutation, and selection. Some looks more organized, tied to developmental programs and loss of cell-identity control. Some may become locally causal by altering transcriptional state, stem-cell behavior, repair capacity, or malignant risk. What the field still lacks is proof that methylation drift, taken in the aggregate, is the primary upstream engine of organismal aging rather than a mixed record of several upstream processes.
Core thesis: DNA methylation drift should not be treated as either a passive stain or a master cause. It is better read as a composite aging layer. Part of it records stochastic and clonal history. Part of it appears functionally involved in cell-state instability and tissue decline. The strongest current position is that methylation drift is partly consequence, partly participant, and still not a complete causal theory of aging by itself.
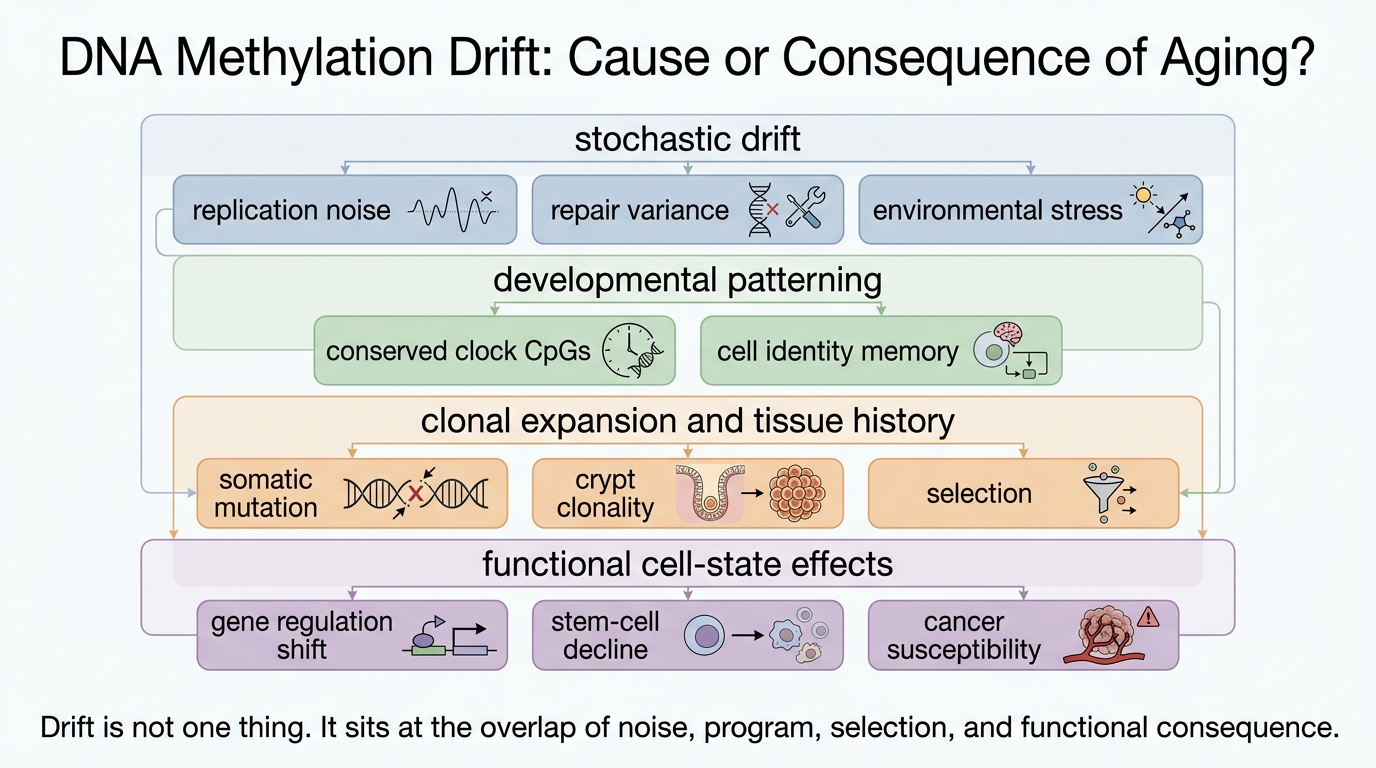
Why Methylation Drift Became Central
Methylation marks became central because they solved a measurement problem before they solved a mechanism problem. Horvath's 2013 clock showed that DNA methylation patterns can estimate age across many tissues and cell types with unusual accuracy. That result established that age leaves a consistent epigenetic footprint. It did not establish why those specific CpGs move, or whether moving them back would by itself reverse the biology that made them move.
The distinction matters. Biomarkers can be predictive without being primary drivers. Blood pressure predicts vascular risk but is not the whole disease architecture. Glycated hemoglobin predicts metabolic control but is not the entire mechanism of diabetes. Methylation clocks may occupy a similar category in aging: unusually useful integrators of biological wear, developmental memory, and tissue context, but not necessarily the single master layer from which all aging decline flows.
That said, methylation biology is too integrated with gene regulation to dismiss as a mere readout. Methylation marks help maintain cell identity, constrain aberrant expression programs, and encode parts of chromatin state. If those controls drift far enough, the result need not be neutral. The real question is not whether methylation matters. The real question is how much of age-linked methylation change is causally upstream, and at what biological scale.
The Strongest Evidence That Drift Reflects Aging
The strongest evidence for consequence comes from the breadth and heterogeneity of the signal. Recent single-cell work from Gladyshev's group showed that epigenetic aging includes a major stochastic component rather than one smooth deterministic program. That matters because stochastic accumulation is exactly what one expects if cells are recording many local histories across replication, damage, repair, and environmental stress. The picture looks less like a single aging switch and more like distributed maintenance failure.
Work on tissue-specific drift reinforces that conclusion. In 2025, Koch and colleagues argued that part of the clock signal may be explained by somatic mutation processes that interact with methylated cytosines. In the intestine, Krepelova and colleagues found that aging-associated methylation drift expands with clonality and crypt dynamics. Those findings do not reduce methylation drift to random garbage. They do show that at least part of the signal is downstream of other aging processes such as mutation burden, stem-cell competition, and tissue architecture.
The consequence view is therefore well supported in one narrow sense: methylation drift captures what aging has already done to cells. It records maintenance history. It records lineage history. It records the fact that old tissues are mosaics of cells with different experiences and different repair outcomes.
The Strongest Evidence That Drift Might Also Participate
The argument for participation becomes stronger when drift is tied to loss of cell identity or to interventions that reset age-linked methylation patterns while improving phenotype. Human and mammalian clock work has repeatedly shown that methylation age tracks developmental state, and that induced pluripotency can drive age estimates toward zero. More important for an aging article, partial reprogramming in mice has been associated with reversal of clock measures alongside tissue-level physiological benefit.
Those findings do not prove that methylation drift is the root cause. They do show that age-linked epigenetic state is not fixed residue. Some of it is reversible, and that reversibility can travel with improved tissue function. If a signal can be reset in parallel with functional improvement, then dismissing it as merely decorative becomes harder. The remaining uncertainty is directionality. Reprogramming changes many layers at once: transcription, chromatin accessibility, stress response, metabolism, and repair signaling. Methylation reset may be a mediator, a passenger, or both.
The more conservative reading is that methylation drift can become locally causal without being globally sovereign. A promoter or enhancer state that drifts can alter expression of genes relevant to inflammation, differentiation, or stress handling. A stem-cell compartment that loses epigenetic precision may become less regenerative or more malignancy-prone. Those are plausible causal roles. They still do not imply that the whole clock is a single actionable cause of aging.
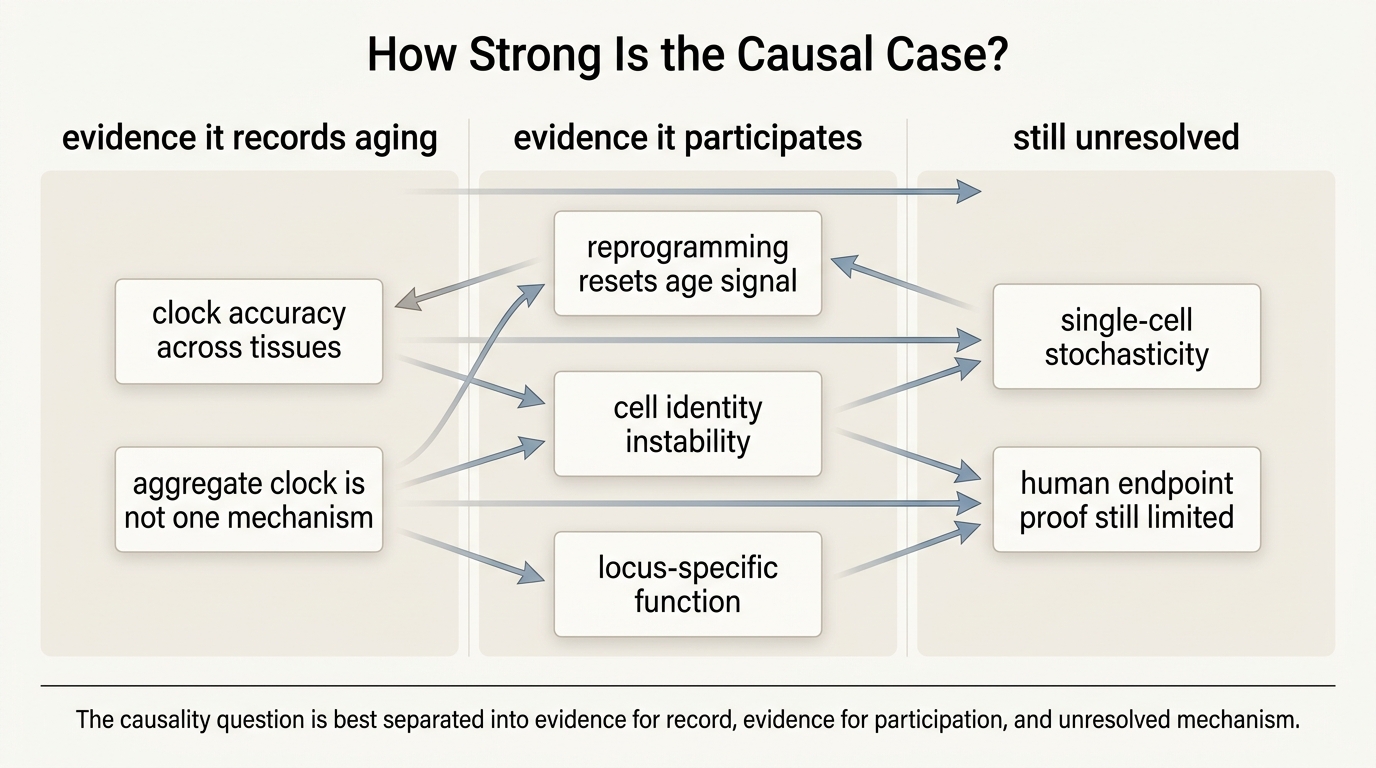
Why The Question Gets Overstated
The public version of this debate usually forces a false choice. Either methylation drift causes aging, or it is just a biomarker. Real systems biology is rarely that binary. A tissue can show age-linked methylation drift because upstream damage and cell turnover are occurring, while those same methylation changes then feed forward into weaker repair, noisier identity, or easier neoplastic transition. Cause and consequence can coexist at different levels of the same system.
The clock industry and parts of the rejuvenation market often flatten this complexity because clear narratives sell better than layered ones. If drift is a master cause, then resetting it sounds like a master treatment. If drift is only a passenger, then clock tools sound less consequential. The research literature is more disciplined. It increasingly points toward mixed architecture: stochastic change, conserved developmental structure, tissue-specific selection, and partially reversible epigenetic state all interacting rather than lining up in one tidy chain.
This is also why the word drift can be misleading. Some methylation changes are not merely random scatter. Mammalian conservation work shows that parts of epigenetic aging are structured across species. That finding argues against the view that all drift is meaningless noise. The field is observing both regularity and randomness at once, which is precisely why the debate remains open.
What Would Strengthen The Causal Case
- Site-specific intervention evidence. Direct editing of age-linked methylation states that improves tissue function without broad reprogramming confounders.
- Tissue-priority mapping. Better evidence for which tissues suffer functional decline from methylation drift first, and which changes are only correlated.
- Clock decomposition. Separation of CpGs that mainly record exposure history from CpGs that mediate cell-state instability or disease risk.
- Longitudinal human intervention data. Durable trials showing that epigenetic-age movement aligns with meaningful health outcomes rather than short biomarker shifts alone.
- Mutation and clonality integration. Stronger models distinguishing methylation change caused by somatic history from methylation change that feeds back into tissue deterioration.
Until those steps are clearer, the most defensible claim remains bounded. Methylation drift is a serious mechanistic layer and a high-value biomarker layer. It is not yet proven to be the single upstream controller that some commercial narratives imply.
Known, Inferred, And Unknown
| Category | Assessment |
|---|---|
| Known | Age-linked DNA methylation patterns support highly predictive epigenetic clocks across tissues and species, which means aging leaves a regular methylation signature. |
| Known | Single-cell and tissue studies show that methylation aging includes major stochastic and clonal components rather than only one deterministic program. |
| Known | Partial reprogramming and pluripotency-related interventions can reset methylation-age signals alongside broader molecular and phenotypic change in model systems. |
| Inferred | Some methylation drift likely participates functionally in loss of cell identity, regenerative decline, or malignant risk even if much of the signal also records upstream damage history. |
| Unknown | How much of the aggregate methylation-clock signal is directly causal for human aging outcomes, and which specific loci or modules are the most actionable. |
The Practical Reading For 2026
DNA methylation drift should be treated as one of the most informative windows into aging, but not as a license for monocausal thinking. It is too biologically integrated to dismiss and too mechanistically unresolved to crown as the singular driver. The right LifeMeter posture is to use methylation drift as a control layer: a way to measure age-linked system state, test interventions more rigorously, and identify where biology may still be reversible.
The field advances when readers resist both simplifications. Drift is not just passive residue. Drift is not yet a complete explanation of aging. It is a dense intersection point where history, identity, damage, and repair become measurable on the same molecular canvas.
Further Reading Inside The Site
This article connects directly to Epigenetic Reprogramming Enters Human Trials, Biological Age Clocks as Decision Tools, and Yamanaka Factors in Vivo: Progress and Constraints. Together they show why clock precision, reprogramming promise, and causality discipline have to stay separate.
Source List
Horvath S. DNA Methylation Age of Human Tissues and Cell Types. Genome Biology. 2013.
Lu AT, Fei Z, Haghani A, et al. Universal DNA Methylation Age Across Mammalian Tissues. Nature Aging. 2023.
Tarkhov AE, Lindstrom-Vautrin T, Zhang S, et al. Nature of Epigenetic Aging From a Single-Cell Perspective. Nature Aging. 2024.
Koch Z, Li A, Evans DS, et al. Somatic Mutation as an Explanation for Epigenetic Aging. Nature Aging. 2025.
Krepelova A, Rasa M, Annunziata F, et al. Iron Homeostasis and Cell Clonality Drive Cancer-Associated Intestinal DNA Methylation Drift in Aging. Nature Aging. 2025.
Browder KC, Reddy P, Yamamoto M, et al. In Vivo Partial Reprogramming Alters Age-Associated Molecular Changes During Physiological Aging in Mice. Nature Aging. 2022.
Translate this longevity claim into a capital-runway decision.
Life extension logic only matters if the balance sheet can carry it. Move into WealthMeter to compare assets, spending, and yield assumptions against the same long-horizon planning problem.