Epigenetic Noise vs Programmed Aging
Aging leaves a reproducible mark on methylation patterns, chromatin state, and transcriptional control. The dispute is over interpretation. Does that pattern mean aging is driven by a coordinated program, or does it show a maintenance system slowly losing precision and accumulating noise?
The strongest answer is less theatrical than either camp prefers. Aging does not look like pure noise, because age-linked epigenetic change is structured enough to support clocks, tissue signatures, and partially reversible states. It does not look like a single clean program either, because those signatures are entangled with damage, repair failure, inflammation, replication history, and metabolic stress.
Established fact: age is associated with reproducible methylation drift, chromatin remodeling, and shifts in gene expression that track biological age across tissues. Reasoned inference: those changes are best read as a layered interaction in which imperfect maintenance creates noise, tissue-state responses channel part of that noise into structured patterns, and some of those patterns then reinforce functional decline.
Why This Argument Exists At All
The argument exists because the data support two partially conflicting impressions. On one side, epigenetic clocks are strikingly orderly. A large set of methylation sites moves with age in ways that are stable enough to estimate chronological age, biological age, mortality risk, and intervention response. That regularity tempts observers to describe aging as a programmed process.
On the other side, the same aging tissues show mounting heterogeneity, transcriptional instability, clonal divergence, inflammatory distortion, and evidence of declining repair quality. That looks less like a precise program and more like a control system losing fidelity. A machine can drift in patterned ways without being designed to fail on schedule. A reproducible breakdown is still a breakdown.
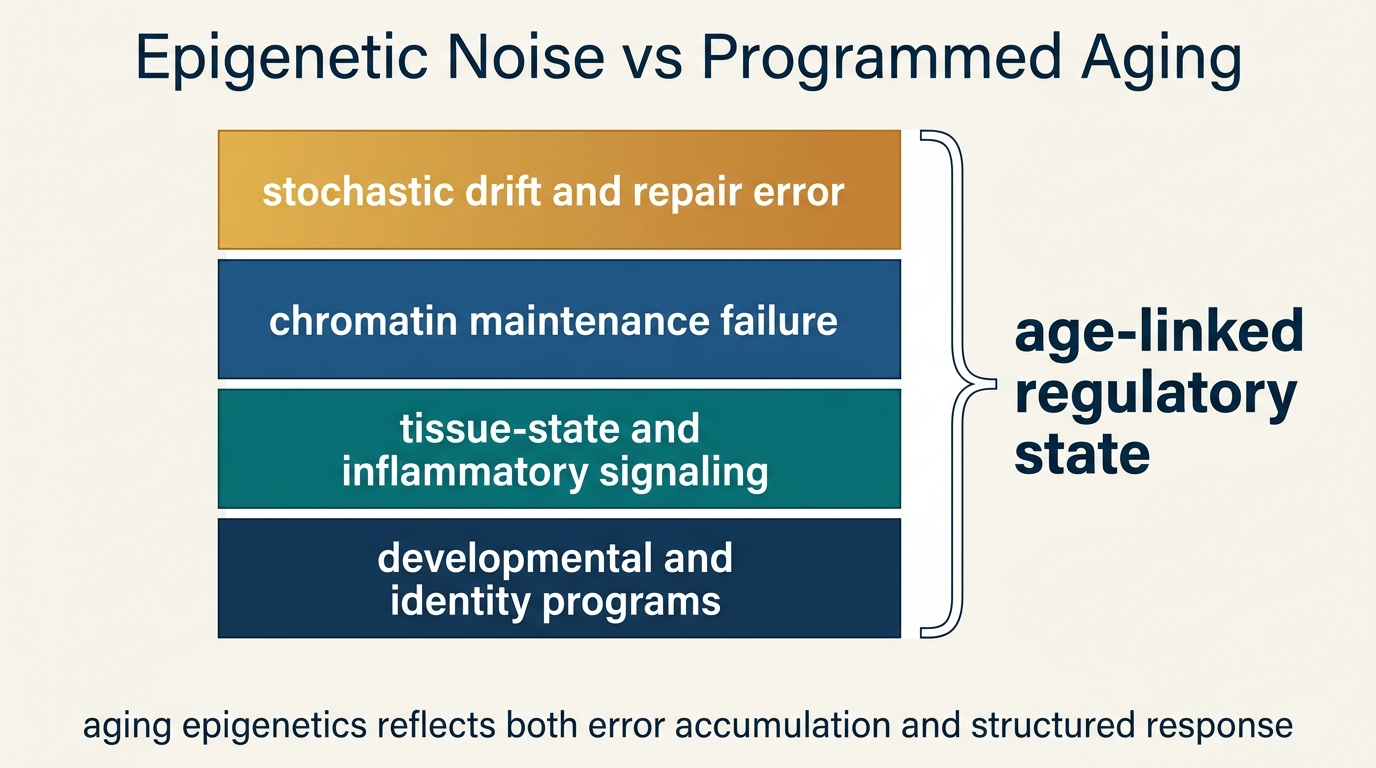
What Epigenetic Noise Actually Means
Epigenetic noise does not mean random marks are sprayed across the genome with no structure. It refers to declining precision in how chromatin state is maintained, copied, and repaired over time. DNA damage responses, replication stress, oxidative injury, metabolic imbalance, and altered chromatin-binding proteins can all nudge regulatory marks away from prior states. Once enough of those nudges accumulate, cells begin to express the same genome through a less reliable regulatory map.
This frame aligns with the broader maintenance view of aging. A young cell preserves identity by continuously policing which genes stay open, which stay closed, and which respond only to specific cues. Age weakens that policing. The result is not chaos in every locus. It is a rising error rate in a system that depends on disciplined state control.
Why The Programmed-Aging View Persists
The programmed-aging view persists because some age-linked epigenetic changes are coordinated, tissue aware, and directionally consistent. Development leaves durable regulatory scaffolds. Endocrine signaling, nutrient sensing, inflammatory tone, and stress-response networks can all produce organized shifts in chromatin and transcription. Some of those shifts may be adaptive early and costly later. That keeps the door open to quasi-programmed aging logic.
Still, the existence of coordinated age-linked regulation does not prove that organisms carry a dedicated aging program in the strong sense. It may simply show that long-lived tissues respond to cumulative damage through recurrent pathways, and those pathways look structured because biology reuses the same regulators repeatedly. Repeated response should not be confused with intentional senescence of the whole organism.
| Interpretive frame | What it explains well | Where it weakens |
|---|---|---|
| Noise-first model | Heterogeneity, stochastic drift, identity loss, and the interaction with damage accumulation | Understates how reproducible clocks and coordinated tissue signatures can be |
| Program-first model | Regular age-linked trajectories, developmental echoes, and structured transcriptional shifts | Overstates intentionality and can misread damage response as design |
| Layered interaction model | Explains why aging can be both reproducible and progressively disordered | Harder to reduce to one decisive mechanism or therapy story |
Clocks Strengthen The Case For Structure, Not For Simplicity
Epigenetic clocks changed this debate because they showed that age-linked marks are not merely diffuse background damage. If methylation state can estimate aging burden this well, then at least part of the system is structured. That matters. It suggests aging biology leaves a readable regulatory footprint rather than a meaningless haze.
What clocks do not prove is causality. A clock can be predictive without being a master driver. It can summarize the downstream consequences of replication history, inflammation, mitochondrial stress, cell turnover, and repair burden. That is why DNA Methylation Drift: Cause or Consequence of Aging? remains the right companion question. Measurement power and mechanistic primacy are not the same claim.
Partial Reprogramming Is The Strongest Evidence Against A Pure Noise Story
If aging were only random epigenetic corruption, partial reprogramming should not work as well as it does in selected animal systems. Yet some reprogramming experiments can reset parts of the epigenetic and functional phenotype without fully erasing cell identity. That implies the aging state is not just debris. At least part of it is organized enough to be partially reversed.
That does not rescue the strongest programmed-aging thesis either. Reprogramming success may show that cells retain access to younger regulatory attractors, not that aging was prewritten as a clean developmental script. It may simply show that a damaged control system can be pushed back toward a more stable configuration. That is still a maintenance story, just one with a reversible state component. The evidence from partial cellular reprogramming is therefore real, but narrower than slogans about a hidden age program suggest.

Tissue Context Matters More Than The Debate Usually Admits
Epigenetic aging is not uniform. Fast-turnover tissues, stem-cell compartments, immune populations, and long-lived postmitotic cells age through different mixtures of replication history, damage exposure, metabolic state, and local inflammatory pressure. A methylation pattern that looks adaptive in one context may be pathological in another. This is one reason whole-body claims about the meaning of drift often outrun the underlying biology.
The practical implication is that aging should be modeled as a hierarchy of regulatory failures and compensations, not as one global chromatin narrative. The brain, hematopoietic system, liver, and muscle do not sit at the same point on the same path. They share some regulatory motifs, but they age through distinct burdens and reserve constraints.
What This Means For Intervention Logic
The intervention lesson is restrictive. If aging epigenetics were a single program, one master reset might be plausible. If it were only noise, intervention would mostly mean slowing damage and preserving maintenance. The existing evidence points between those poles. Some states may be resettable. Others may depend on preventing ongoing metabolic, inflammatory, or genomic injury from recreating the same drift.
- Treat clocks as measurement tools before treating them as therapeutic maps.
- Separate reversible regulatory state from irreversible structural damage.
- Expect tissue-specific reset windows, not one organism-wide epigenetic correction.
- Judge reprogramming claims by preserved function and safety, not by age-reversal rhetoric.
This is why the cleanest synthesis sits alongside the hallmarks framework and the newer work on biological age clocks. Epigenetic drift matters because it captures system state. It becomes dangerous when readers treat that state readout as proof that aging is either pure entropy or pure design.
Known, Inferred, And Unknown
| Category | Assessment |
|---|---|
| Known | Aging is associated with reproducible shifts in DNA methylation, chromatin organization, and gene regulation across multiple tissues. |
| Known | Epigenetic clocks provide strong evidence that age-linked regulatory change is structured enough to be measured with meaningful predictive power. |
| Known | Partial reprogramming studies show that selected aging-associated epigenetic and functional states can be pushed toward a younger profile in animal systems. |
| Inferred | The most defensible model is a layered one in which maintenance failure creates drift, biological response pathways impose structure on that drift, and some structured states later help sustain aging phenotypes. |
| Unknown | Which age-linked epigenetic changes are primary drivers, which are adaptive responses, and which can be reset safely in humans without identity loss, tumor risk, or unstable tissue effects. |
Source Frame
This analysis draws on durable field structure rather than one headline: methylation-drift literature, chromatin-aging reviews, epigenetic-clock work, and partial reprogramming studies that test how reversible age-linked regulatory state may be.
The unresolved question is not whether aging has an epigenetic signature. It is which parts of that signature are merely descriptive, which are causal, and which can be modified without rebuilding the same instability through ongoing damage and tissue stress.
Translate this longevity claim into a capital-runway decision.
Life extension logic only matters if the balance sheet can carry it. Move into WealthMeter to compare assets, spending, and yield assumptions against the same long-horizon planning problem.