Mitochondrial Decline as the Central Aging Driver
People feel aging as a loss of energy long before they describe it in molecular terms. That is one reason mitochondria keep returning to the center of the conversation. When tissues lose energetic flexibility, stress tolerance, and repair capacity, mitochondrial dysfunction is usually somewhere in the chain. The analytical mistake is to turn that recurring appearance into a claim that mitochondria explain all of aging by themselves.
The strongest version of the mitochondrial case is not that mitochondria are a fashionable target. It is that they sit at a convergence point for ATP production, redox control, apoptosis, inflammatory signaling, stem-cell behavior, and stress adaptation. A system with that much responsibility can plausibly amplify damage across multiple tissues. The weaker version of the claim is that mitochondria must therefore be the singular root cause of aging. Current evidence does not justify that compression.
Core thesis: mitochondrial decline is central because it links energy failure, signaling distortion, quality-control breakdown, and tissue-level repair constraints across the aging phenotype. That does not mean mitochondria act alone. It means they are one of the most credible bottleneck layers in a larger maintenance network.
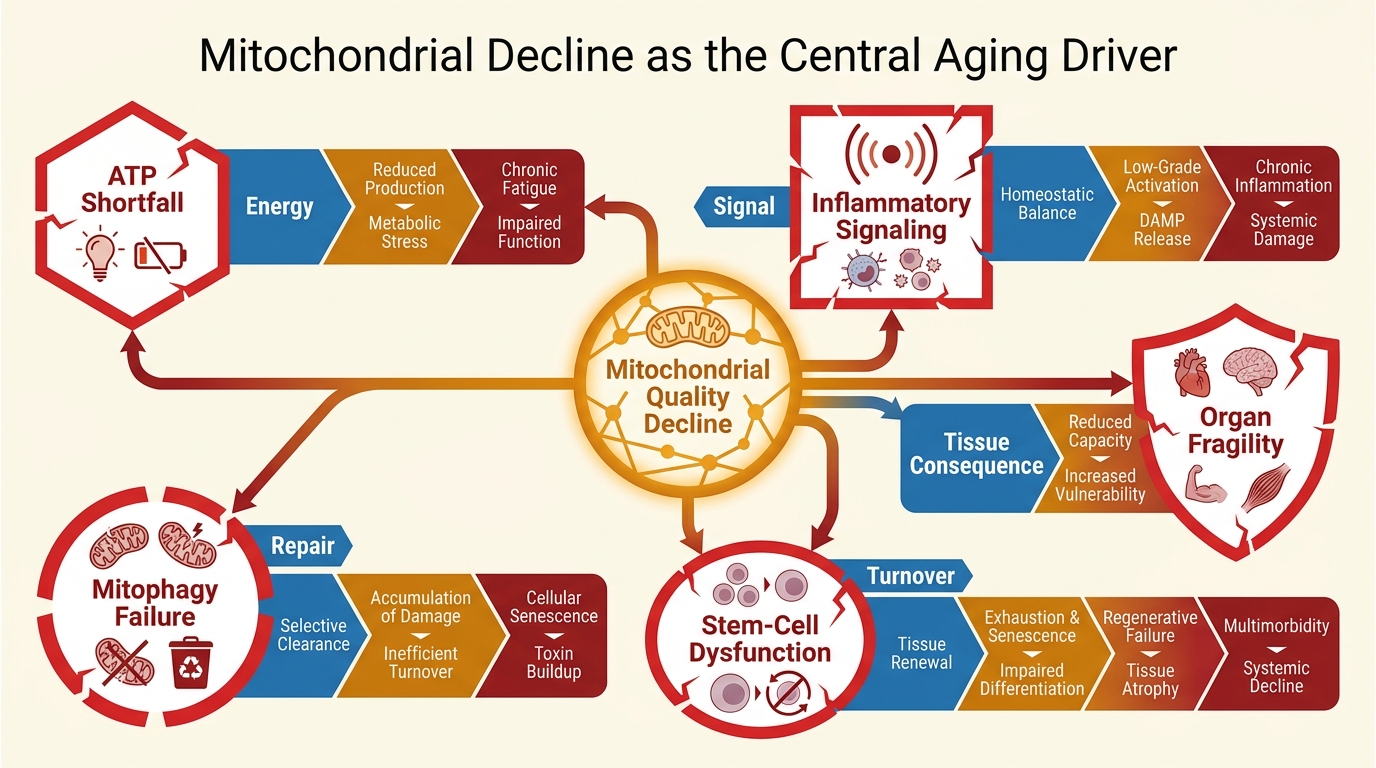
Why Mitochondria Keep Reappearing Across Aging Biology
Mitochondria are not only energy producers. They influence metabolite pools, reactive oxygen species handling, innate immune signaling, calcium buffering, and cell-fate decisions. That breadth matters. A damaged lysosome causes one class of problems. A damaged extracellular matrix causes another. Mitochondrial decline can modify both upstream stress exposure and downstream response capacity across many cell types at once.
This is why mitochondrial decline shows up in multiple hallmark lanes rather than one isolated bucket. It touches proteostasis through folding stress, stem-cell function through niche energy state, senescence through mitochondrial dysfunction-associated inflammatory signaling, and tissue repair through bioenergetic limits. The centrality claim is therefore strongest when it means cross-hallmark reach, not total explanatory monopoly.
Established fact supports that reach. It does not yet prove that mitochondrial decline initiates every major aging program earlier than other damage layers do, or that fixing mitochondria alone would normalize whole-organism aging.
The Case For Calling It Central
There are three defensible reasons to place mitochondria near the center of the map. First, mitochondrial quality falls with age through accumulated damage, impaired biogenesis, altered dynamics, and weaker mitophagy. Second, tissues with high energy demand such as brain, heart, muscle, and kidney are unusually sensitive to that drift. Third, mitochondrial stress can turn into signaling that worsens inflammation, genomic stress response, and cell-state deterioration rather than remaining a private energy problem.
That is what makes the topic more than a fatigue narrative. A mitochondrial lesion can reduce output and simultaneously change what the cell believes about its own survival state. Once that happens, compensatory pathways can become chronic, inflammatory, or maladaptive. Aging then looks less like a single broken engine and more like a system that keeps paying higher metabolic costs for weaker biological performance.
Where The Central-Driver Story Overreaches
The overreach starts when a real bottleneck is recast as a universal first cause. Aging also involves extracellular matrix stiffening, stem-cell niche drift, clonal selection, epigenetic drift, proteostatic failure, and organism-level signaling changes that are not reducible to mitochondria alone. In some tissues mitochondrial dysfunction may be early and causal. In others it may be downstream of chronic inflammation, vascular change, DNA damage, or altered nutrient sensing.
This distinction matters for intervention design. If mitochondria are always upstream, then mitochondrial rescue should dominate the field. If mitochondria are often a central relay inside a broader damage network, then mitochondrial therapies may help most when paired with other repair layers. The second reading is more defensible today.
The same caution applies to consumer narratives. Raising NAD levels, taking ketones, or supporting exercise capacity does not automatically mean mitochondrial aging has been durably repaired. Some interventions improve throughput or signaling without reversing the full quality-control problem.
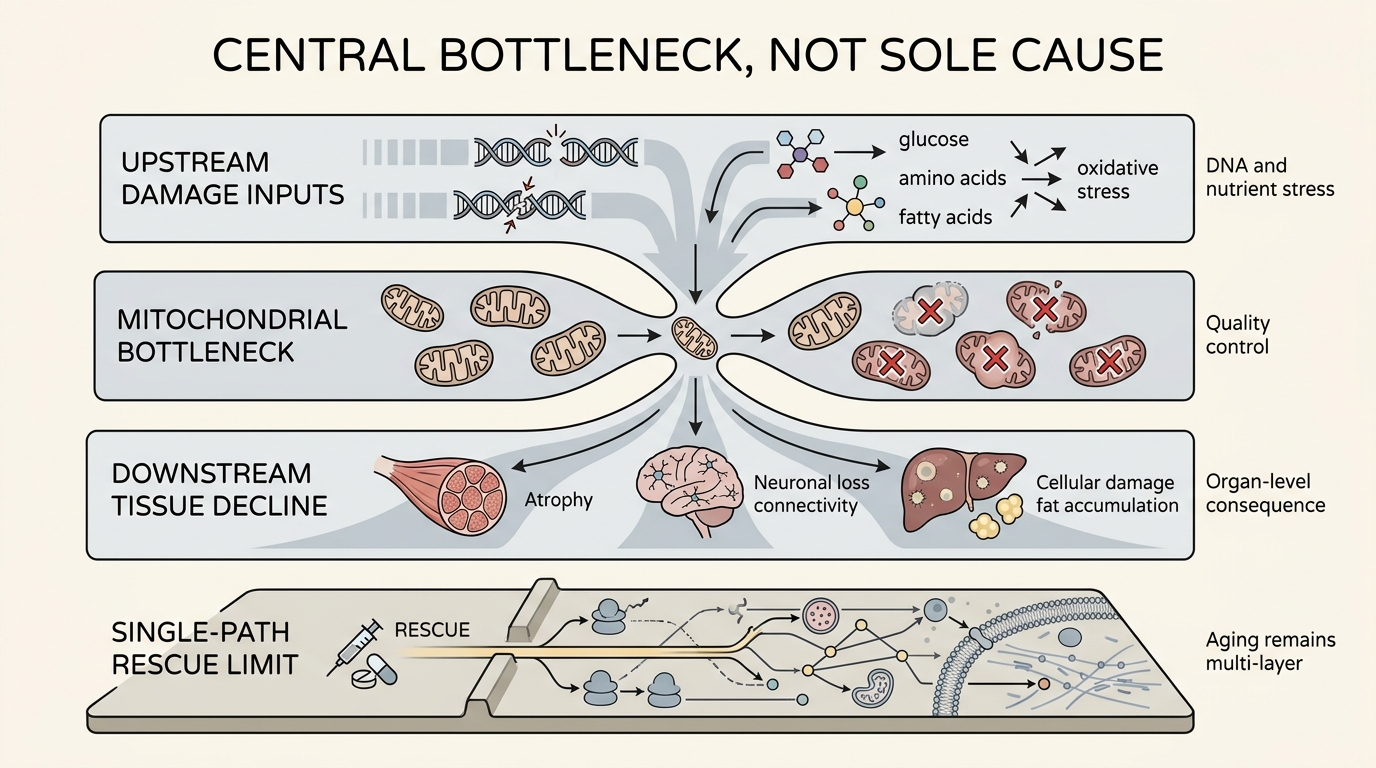
Why Quality Control Matters More Than The Old ROS Story
Older popular explanations often reduced mitochondrial aging to oxidative damage alone. That is too narrow. Reactive oxygen species matter, but the more useful framework tracks mitochondrial turnover, fusion and fission balance, mitophagy quality, genome maintenance, and the cell's ability to keep damaged organelles from dominating the network. A system that cannot clear weak mitochondria eventually pays in both energy output and noisy signaling.
This is one reason the topic connects directly to NAD+ Restoration: Mechanism, Hype, and Clinical Signal and Sirtuins in Humans: From Yeast Biology to Clinical Relevance. Both lines are often marketed as simple mitochondrial rescue stories. The stronger interpretation is narrower: they interact with mitochondrial maintenance and stress response, but their human anti-aging translation still depends on whether they change durable system behavior rather than only shifting one node.
What Would Count As Real Mitochondrial Translation
- Strong evidence: interventions that improve validated functional outcomes in aging humans while also showing coherent changes in mitochondrial quality, not only transient energetic signal.
- Intermediate evidence: durable improvement in disease-relevant tissues where mitochondrial dysfunction is clearly implicated and the mechanism is measured rather than assumed.
- Weak evidence: generalized longevity claims built from biomarker movement, supplement marketing, or exercise-mimetic language without tissue-level durability.
The field still lacks a clean demonstration that one mitochondrial program broadly resets human aging across organs. What it does have is a strong reason to treat mitochondria as a recurring leverage layer that no serious aging model can ignore.
Known, Inferred, And Unknown
| Category | Assessment |
|---|---|
| Known | Mitochondrial function declines with age across multiple tissues through altered turnover, impaired bioenergetics, and weaker organelle quality control. |
| Known | Mitochondria influence energy production, inflammatory signaling, apoptosis, and stress adaptation, which gives their decline system-level consequences. |
| Known | Current human evidence does not prove that mitochondrial rescue alone can broadly reverse organismal aging. |
| Inferred | Mitochondria function best as a central-bottleneck concept inside a layered maintenance model rather than as the lone master cause of aging. |
| Unknown | Which combination of biogenesis support, mitophagy improvement, metabolic rewiring, and tissue targeting can produce durable multi-organ human benefit. |
The Practical Reading For LifeMeter
If an intervention claims to slow aging through mitochondria, the right question is not whether mitochondria matter. They do. The right question is what exactly changed: energetic throughput, organelle turnover, inflammatory spillover, tissue function, or only a proxy marker that sounds mitochondrial. Once that distinction is enforced, many broad claims shrink quickly.
The stronger strategic view is that mitochondria belong near the center of the longevity map because they connect many failure modes. That makes them a serious target. It does not give them permission to erase the rest of the map. A robust longevity program still has to ask how mitochondrial rescue interacts with matrix damage, senescence burden, stem-cell decline, and whole-body signaling state.
Further Reading Inside The Site
This article connects directly to Hallmarks of Aging Revisited: What Changed Since 2013, NAD+ Restoration: Mechanism, Hype, and Clinical Signal, and Extracellular Matrix Aging: The Forgotten Target. Together they show why one biologically central layer still has to be placed inside a wider systems model.
Source List
López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013.
López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. Hallmarks of Aging: An Expanding Universe. Cell. 2023.
Sun N, Youle RJ, Finkel T. The mitochondrial basis of aging. Molecular Cell. 2016.
Jang JY, Blum A, Liu J, Finkel T. The role of mitochondria in aging. Journal of Clinical Investigation. 2018.
Selected mitochondrial-quality-control and mitophagy literature used for the systems interpretation here.
Translate this longevity claim into a capital-runway decision.
Life extension logic only matters if the balance sheet can carry it. Move into WealthMeter to compare assets, spending, and yield assumptions against the same long-horizon planning problem.