Tissue-Specific Senescence: Why Location Matters
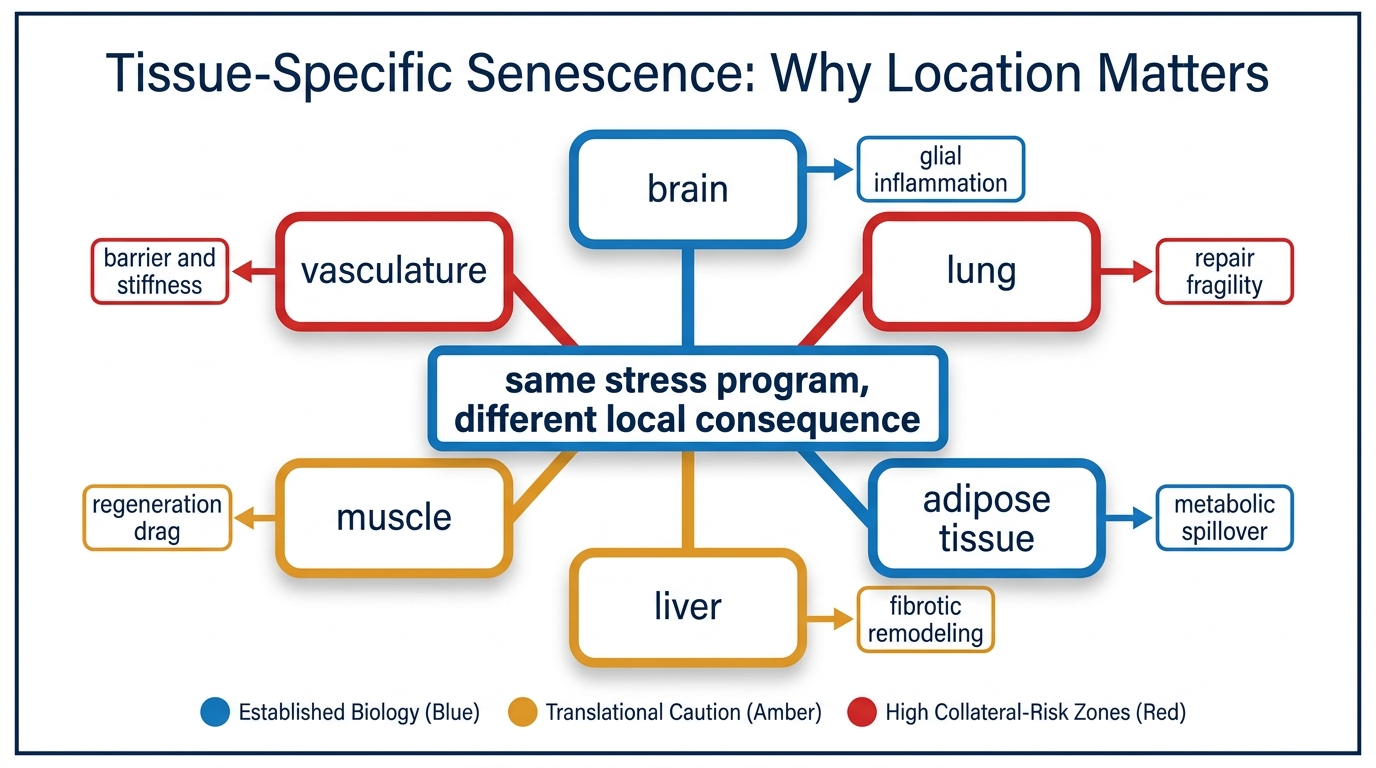
Senescence is often discussed as if one number could summarize it. That framing is too coarse. A senescent endothelial cell, a senescent adipocyte precursor, a senescent stellate cell in the liver, and a senescent glial cell do not create the same biological problem. They do not signal into the same neighborhood, they do not degrade the same function, and they should not be evaluated with the same intervention logic.
The field now has enough evidence to reject a generic senescence story. Senescence is real, harmful when persistent, and relevant to aging. What remains far more conditional is how much a given tissue contributes to whole-organism decline, whether the local senescent program is pathogenic or partly compensatory, and whether clearance, suppression, or environmental repair is the right response. Tissue location is therefore not a side detail. It is the thing that determines meaning.
Core thesis: senescence is a distributed tissue phenomenon rather than one interchangeable burden. The strongest established claim is that persistent senescent states disrupt local repair, inflammatory tone, and structural function in tissue-specific ways. The stronger claim, that one generalized senolytic strategy can improve ordinary human aging across organs, remains unproven because each tissue has different cell types, tolerances, and regeneration constraints.
Why A Single Senescence Score Misleads
Cellular senescence is best described as a family of stress programs rather than one molecular identity. Gorgoulis and colleagues made this point directly when they argued for a more disciplined path forward: marker panels vary, triggers vary, and consequences vary. That heterogeneity matters more in aging than in cell culture because whole tissues have their own repair logic, immune surveillance quality, extracellular matrix structure, and turnover speed.
A senescent burden estimate therefore always hides an important second question. Burden where? A modest accumulation in a tissue with low regenerative reserve may matter more than a larger burden in a compartment that turns over faster or tolerates transient damage better. The useful metric is not simply cell count. It is cell count multiplied by local function loss, local inflammatory spillover, and the tissue's ability to recover after intervention.
This is why readers should be skeptical when one biomarker or one broad animal result is used to imply a universal anti-aging pathway. Senescence biology is shared at the level of stress responses and SASP signaling, but the consequences are still filtered through organ architecture.
Adipose Tissue Is Small In Cell Count And Large In Systemic Consequence
Adipose tissue is one of the clearest examples of why location matters. Senescent adipocyte progenitors and stromal cells do not merely age in place. They can alter adipogenesis, insulin sensitivity, inflammatory signaling, and the quality of metabolic overflow into liver and muscle. A relatively localized change can therefore create body-wide metabolic consequences.
This does not mean adipose senescence explains all metabolic aging. It does mean that adipose is a high-leverage compartment because it sits at the junction of nutrient storage, endocrine signaling, macrophage traffic, and ectopic lipid stress. In practical terms, a senescence intervention that improves adipose resilience could matter disproportionately even if total senescent-cell burden elsewhere remains significant.
The inference is strong, but not unlimited. What is established is that dysfunctional adipose biology contributes to systemic aging pressure. What remains unresolved is the exact fraction attributable to senescence versus obesity-related inflammation, mitochondrial dysfunction, fibrosis, and broader immunometabolic drift.
Endothelium And Vasculature Translate Senescence Into Flow Problems
Vascular tissues make the same point from another angle. Senescence in endothelial and smooth-muscle compartments does not mainly matter because of cell count. It matters because the vasculature is a distribution system. Small local impairments can propagate into worse nitric-oxide signaling, stiffer vessels, pro-inflammatory surface behavior, poorer repair after injury, and weaker organ perfusion.
That shifts the practical question. In vascular aging, readers should ask less about whether senescence exists and more about which functional output is breaking first: vasodilation, barrier integrity, thrombosis control, or structural stiffness. A senolytic that removes damaged cells but destabilizes an already fragile endothelial layer could be worse than a strategy that suppresses inflammatory output while preserving structural continuity.
This is one reason senomorphics remain relevant in the discussion. In some tissues, the correct goal may not be maximal clearance. It may be narrower phenotype control while other repair pathways are strengthened.
Fibrotic Organs Carry A Different Tradeoff
Liver, lung, and kidney tissue illustrate a harder version of the same problem. In these organs, senescent states can contribute to fibrosis, distorted wound-healing, and chronic inflammatory signaling. At the same time, senescence can also act as a transient brake on damaged-cell proliferation during injury responses. Clearing cells indiscriminately in a fibrotic organ is therefore not obviously equivalent to improving it.
The early human idiopathic pulmonary fibrosis senolytic pilot is useful here not because it resolved the field, but because it showed how cautious translation must be. A fragile fibrotic organ is exactly where readers should expect the largest gap between mechanistic appeal and dependable clinical outcome. Tissue architecture, timing, and repair reserve all matter.
The practical lesson is that fibrotic tissue raises the bar for intervention design. It is not enough to show that a senescent-cell population is present. The intervention has to improve the tissue's future operating state rather than merely change one histologic snapshot.
Brain Senescence Is Not A Copy Of Peripheral Senescence
Brain aging adds another layer of constraint. Senescent-like states in astrocytes, microglia, oligodendrocyte-lineage cells, and vascular support cells sit inside a compartment with limited regeneration, tight immune access rules, and heavy dependence on local signaling balance. A modest shift in glial inflammatory tone or blood-brain barrier support can matter more than a crude tissue average implies.
That makes the brain a poor fit for simplistic clearance narratives. The central nervous system is not only another target organ. It is a protected signaling environment. Delivery, off-target inflammation, and the distinction between adaptive reactivity and harmful persistence all matter more here than in a more replaceable peripheral tissue.
For readers following neurodegeneration claims, the disciplined position is clear. Senescence-related mechanisms likely matter in the brain, but intervention confidence should remain lower than in some peripheral tissues until cell identity, timing, and safety are more sharply resolved.

Why Intervention Logic Has To Follow The Tissue
The main translational mistake in this field is to assume that target validation and intervention design are the same thing. They are not. A tissue can show clear senescence involvement while still requiring a different therapeutic move than another tissue with the same broad markers. Clearance may be correct where persistent inflammatory secretion dominates and replacement capacity is adequate. Suppression may be safer where cells are dysfunctional but still structurally entangled with a fragile tissue. Upstream repair may matter more where senescence is secondary to ongoing mitochondrial, matrix, or immune failure.
This is why tissue-specific senescence should be read alongside immune surveillance, extracellular matrix aging, and stem-cell reserve. Senescence is rarely the only failing layer. It is usually one part of a local deterioration stack. Readers who understand that stack are much less likely to overread a single intervention headline.
Known, Inferred, And Unknown
| Category | Assessment |
|---|---|
| Known | Cellular senescence is heterogeneous across triggers, markers, and functional outputs, which makes tissue context essential for interpretation. |
| Known | Persistent senescent states can disrupt local tissue biology through inflammatory signaling, altered repair, and structural dysfunction, but the dominant consequence differs by organ. |
| Known | Adipose, vascular, fibrotic, and neural compartments each present different senescence-related risks and different tolerance for aggressive clearance. |
| Inferred | Some of the strongest future gains in senescence medicine will likely come from matching intervention class to tissue logic rather than from maximizing whole-body clearance. |
| Unknown | Which human tissues contribute most to age-related decline at realistic senescent-cell burdens, and which treatment sequences improve long-run function without impairing repair or regeneration. |
The Practical Reading For LifeMeter
The useful takeaway is simple: stop treating senescence as a monolith. Serious readers should ask which tissue is being discussed, which cell populations are implicated, what local function is failing, and whether the proposed intervention matches that failure mode. That is the difference between mechanistic literacy and slogan consumption.
This is also why broad anti-aging promises around senolytics remain premature. The field has a credible target class, real preclinical depth, and real translational momentum. It does not yet have a universal human answer, because no such universal tissue exists. Aging happens in organs, niches, barriers, and repair systems. Senescence medicine has to meet it there.
Further Reading Inside The Site
This article connects directly to Senolytics: From Mouse Lifespan Gains to Human Trials, Senomorphics vs Senolytics: Slowing vs Removing Damage, Immune System Aging and Senescent Cell Accumulation, and Extracellular Matrix Aging: The Forgotten Target. Together they show why senescence has to be read as part of a local maintenance system rather than as a standalone villain.
Source List
Gorgoulis V, Adams PD, Alimonti A, et al. Cellular Senescence: Defining a Path Forward. Cell. 2019.
Hernandez-Segura A, Nehme J, Demaria M. Hallmarks of Cellular Senescence. Trends in Cell Biology. 2018.
Childs BG, Durik M, Baker DJ, van Deursen JM. Cellular Senescence in Aging and Age-Related Disease. Nature Medicine. 2015.
Kirkland JL, Tchkonia T. Senolytic Drugs: From Discovery to Translation. Journal of Internal Medicine. 2020.
Tuttle CSL, Waaijer MEC, Slee-Valentijn M, et al. Cellular Senescence and Chronological Age in Various Human Tissues: A Systematic Review and Meta-Analysis. Aging Cell. 2020.
Justice JN, Nambiar AM, Tchkonia T, et al. Senolytics in Idiopathic Pulmonary Fibrosis: Results From a First-in-Human, Open-Label, Pilot Study. EBioMedicine. 2019.
Translate this longevity claim into a capital-runway decision.
Life extension logic only matters if the balance sheet can carry it. Move into WealthMeter to compare assets, spending, and yield assumptions against the same long-horizon planning problem.