Immune System Aging and Senescent Cell Accumulation
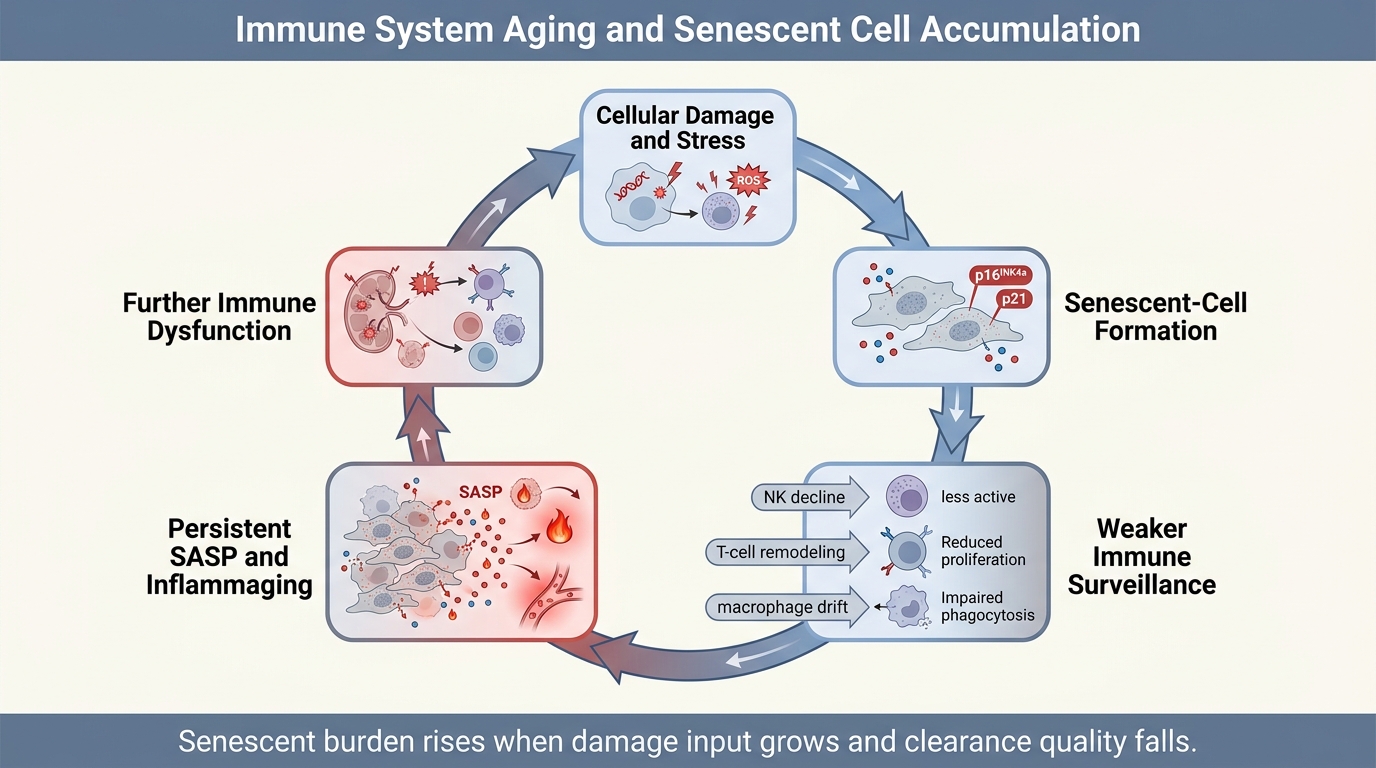
Aging tissues do not accumulate senescent cells only because more cells are damaged. They also accumulate them because the clearance system ages. The immune system is part janitor, part referee, and part repair coordinator in senescence biology. When that system loses precision, senescent cells persist longer, inflammatory signaling spreads farther, and tissue recovery becomes less reliable.
That framing matters because public senescence discussions often isolate the damaged cell and ignore the surrounding control network. The field does not support that simplification. Senescent burden reflects production, recognition, clearance, and tissue context. Immune aging changes all four. The result is not a single switch but a feedback loop in which weaker surveillance allows more persistent senescence, and persistent senescence further degrades immune performance through chronic inflammatory stress.
Core thesis: immunosenescence helps drive senescent-cell accumulation because aging weakens the immune system's ability to recognize, contain, and clear senescent cells across tissues. The strongest evidence supports a reciprocal loop: aging damage creates senescence, senescent cells intensify inflammatory burden, and the aging immune system becomes progressively less able to resolve that burden. What remains unresolved is how much of that loop is reversible in humans, by which interventions, and in which tissues.
Why Senescent Cells Need An Immune Context
Cellular senescence is best understood as a stress response with both protective and harmful faces. It can suppress replication of damaged cells, aid wound-healing transitions, and limit some oncogenic threats. The problem begins when senescent cells persist and their secretory phenotype remains active after the useful window has passed. Hernandez-Segura, Nehme, and Demaria emphasized this point clearly: senescence is not one molecular identity but a context-dependent state whose consequences depend heavily on duration and environment.
The immune system is part of that environment. Senescent cells signal for recognition through cytokines, chemokines, and altered surface programs. Immune cells then decide whether the tissue will resolve the state, tolerate it, or drift into chronic dysfunction. When that resolution pathway weakens, the same senescence program that can be transiently adaptive becomes chronically harmful.
This is why accumulation should not be described as passive clutter. It is evidence that a maintenance process has slowed or failed.
What Immune Aging Changes
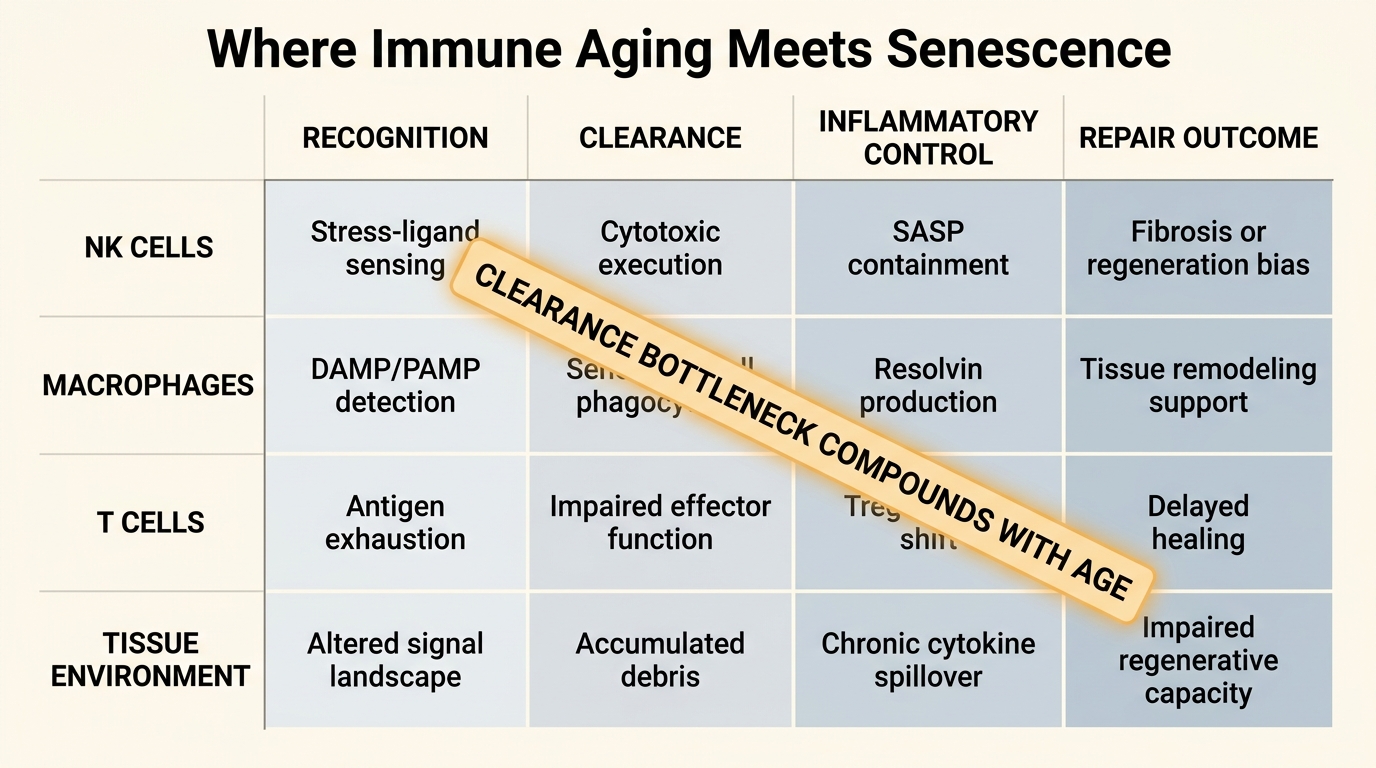
Immunosenescence is not merely lower immunity. It is a reconfiguration of the immune system that includes weaker naive-cell reserves, altered innate signaling, narrower receptor diversity, impaired cytotoxic response, and a higher baseline of chronic inflammatory tone. Nikolich-Zugich's review remains useful because it separates simple decline from remodeling. Some compartments lose function outright, some become biased toward memory and inflammatory persistence, and some remain active but less well targeted.
That matters for senescent-cell control because clearance depends on precision. Natural killer cells, macrophages, and T-cell populations do not simply need to be present. They need to detect stressed cells accurately, move into tissue, execute clearance, and then terminate the response without excess collateral injury. Aging makes each step less reliable. The result is not universal failure in every tissue, but a higher probability that senescent cells remain in place long enough to reshape the local environment.
Majewska and Krizhanovsky describe this as a surveillance problem. Senescent cells and immune cells are in constant dialogue. Aging distorts both sides of that dialogue. Senescent cells may become harder to clear, and immune cells become less capable of interpreting and acting on the signal.
Why Senescent Burden Rises With Age
Human tissue data support the broad accumulation story. Tuttle and colleagues' systematic review found that senescence-associated markers generally increase with age across human tissues, although marker choice and tissue context complicate the exact rate. That is a measured observation, not only a mouse inference. Senescent burden rises in real human biology.
The harder question is why it rises. Part of the answer is straightforward: older tissues experience more replication history, more DNA damage, more mitochondrial stress, and more inflammatory or fibrotic injury. But the rise in burden does not follow from damage input alone. A stable system could still clear a significant share of those cells. The evidence increasingly suggests that aging weakens that output channel.
This is where immune aging and senescent accumulation meet. If production rises and clearance falls at the same time, accumulation becomes structurally likely. A useful way to think about aging tissues is therefore net senescent burden equals generation minus removal, filtered through tissue-specific tolerance.
The Inflammaging Link Is Not Incidental
Chronic low-grade inflammation is one of the best-supported system-level features of aging. Senescent cells contribute to it through the senescence-associated secretory phenotype, and inflammaging in turn reshapes immune behavior in ways that can sustain further senescence. The two processes are not identical, but they overlap heavily.
Ajoolabady and colleagues describe inflammaging and immunosenescence as coupled features of biological aging rather than isolated modules. That is the right framing for LifeMeter readers because it explains why senescence is difficult to treat with one-dimensional logic. Even if a therapy reduces one senescent-cell pool, the tissue may remain inflammatory, fibrotic, metabolically stressed, or poorly surveilled. The surrounding immune environment continues to matter.
This also explains why many strong preclinical senescence interventions look less decisive when translated toward human systems. The target is not only the cell. It is the cell inside an aged network.
What This Means For Senolytics
Senolytic enthusiasm often focuses on removal. That is understandable, but incomplete. If the aged immune system remains impaired, one burst of pharmacologic clearance may not produce durable control. Newly damaged cells can still enter senescence, and the tissue can still fail to manage them. The practical question is therefore not only whether a senolytic kills senescent cells. It is whether the broader surveillance system improves enough to change the future balance.
The early human pilot from Justice and colleagues in idiopathic pulmonary fibrosis showed why readers should remain disciplined. The study supported feasibility and motivated further translation. It did not settle long-horizon human outcomes, tissue durability, or the broader question of which senescent-cell programs are most worth clearing. That restraint still applies.
Newer immune-engineering approaches, including senescence-targeted CAR T work, show why the field is intellectually interesting. They turn the surveillance problem into a therapeutic design problem. But that does not mean the delivery, safety, and tissue-specificity questions are solved. It means the field is beginning to address the right level of mechanism.
Known, Inferred, And Unknown
| Category | Assessment |
|---|---|
| Known | Markers associated with cellular senescence generally rise with age across multiple human tissues, although no single marker captures the entire state cleanly. |
| Known | Immune aging changes surveillance quality through altered innate and adaptive function, weaker cytotoxic precision, and a higher chronic inflammatory baseline. |
| Known | Senescent cells and the aging immune system interact bidirectionally, which helps explain why persistent senescence and inflammaging often travel together. |
| Inferred | A substantial share of rising senescent burden with age reflects not only higher damage input but also weaker immune-mediated clearance and poorer resolution. |
| Unknown | Which human tissues, immune compartments, and intervention sequences can restore durable senescence control without creating unacceptable repair loss, fibrosis risk, or immune toxicity. |
The Practical Reading For LifeMeter
The useful lesson is that senescence should be judged as a systems-maintenance problem, not as a single-cell villain story. A serious longevity strategy has to ask where the aged immune system is failing, whether senescent burden is a primary driver in that tissue, and whether the intervention improves future control rather than only one current snapshot.
This is also why readers should treat sweeping human anti-senescence claims cautiously. The field has real mechanistic credibility, real translational energy, and real limits. The strongest current case is that immune aging and senescent accumulation reinforce each other. The weaker and still unresolved case is that we already know how to break that loop safely across ordinary human aging.
Further Reading Inside The Site
This article connects directly to Senomorphics vs Senolytics: Slowing vs Removing Damage, Senolytics Moving into Clinical Translation, and Stem Cell Exhaustion and Replenishment Strategies. Together they show why repair quality depends on cell state, tissue environment, and the immune system's ability to clear damaged states without destabilizing regeneration.
Source List
Hernandez-Segura A, Nehme J, Demaria M. Hallmarks of Cellular Senescence. Trends in Cell Biology. 2018.
Nikolich-Zugich J. The Twilight of Immunity: Emerging Concepts in Aging of the Immune System. Nature Immunology. 2018.
Tuttle CSL, Waaijer MEC, Slee-Valentijn M, et al. Cellular Senescence and Chronological Age in Various Human Tissues: A Systematic Review and Meta-Analysis. Aging Cell. 2020.
Ajoolabady A, Aghamir SMK, Sayed N, et al. Immunosenescence and Inflammaging in Age-Related Diseases. Ageing Research Reviews. 2024.
Majewska A, Krizhanovsky V. The Immune Surveillance of Senescent Cells in Aging and Disease. Nature Reviews Immunology. 2024.
Justice JN, Nambiar AM, Tchkonia T, et al. Senolytics in Idiopathic Pulmonary Fibrosis: Results From a First-in-Human, Open-Label, Pilot Study. EBioMedicine. 2019.
Translate this longevity claim into a capital-runway decision.
Life extension logic only matters if the balance sheet can carry it. Move into WealthMeter to compare assets, spending, and yield assumptions against the same long-horizon planning problem.