Proteostasis Collapse and the Aging Proteome
Aging cells do not only accumulate damaged DNA, dysfunctional mitochondria, or senescent signaling. They also lose control over the protein world that makes cell function possible in the first place. When protein quality control weakens, the proteome stops behaving like a well-maintained inventory and starts behaving like a backlog.
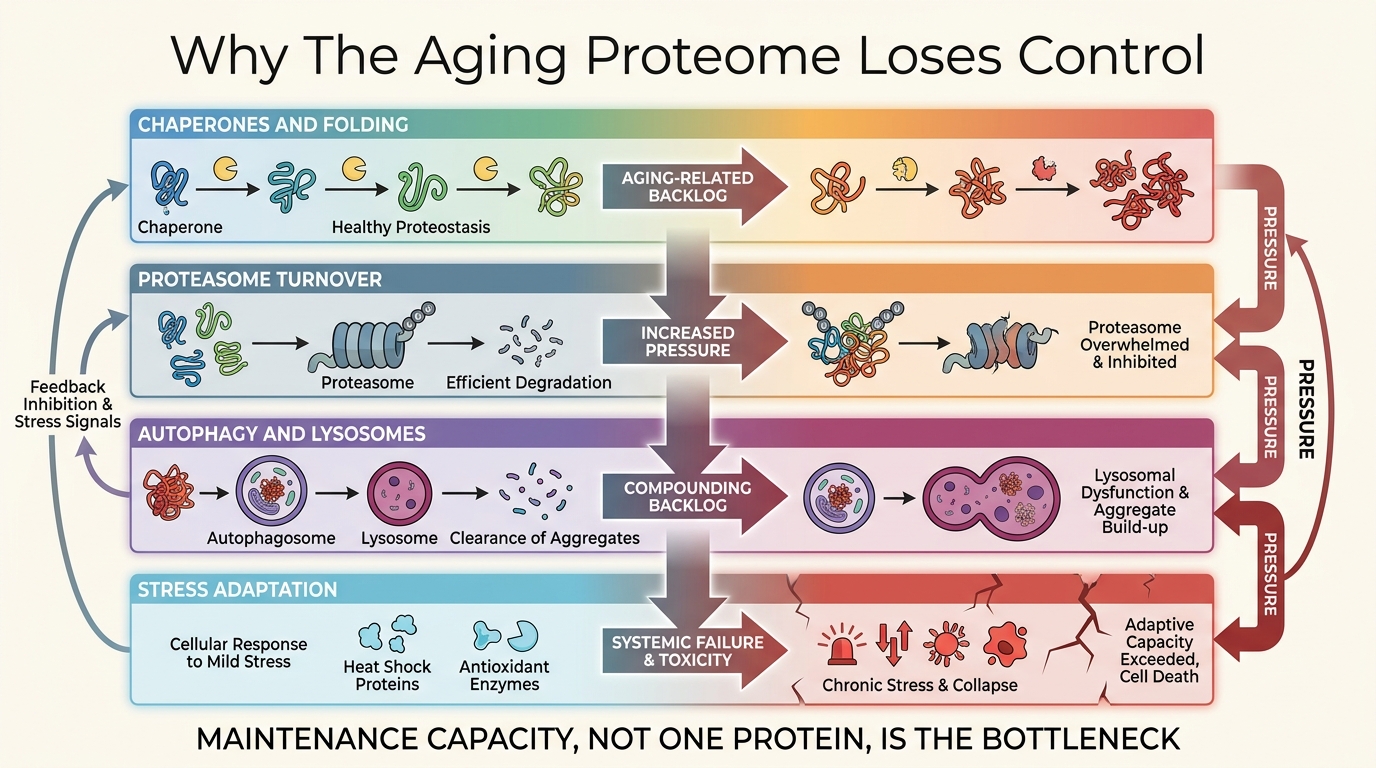
The phrase loss of proteostasis is sometimes treated like shorthand for protein aggregates. That is too narrow. Proteostasis covers synthesis, folding, trafficking, stress response, degradation, and compartment-specific cleanup. The relevant failure is not one bad protein. It is declining capacity across the maintenance stack that keeps damaged proteins from becoming a systems problem.
Established fact: aging is associated with weaker chaperone response, less efficient proteasomal and autophagic clearance, and rising misfolded or damaged protein burden across multiple tissues. Reasoned inference: because those quality-control layers degrade together, proteostasis is best understood as a maintenance bottleneck rather than a single lever waiting for one decisive drug.
Proteostasis Is A Capacity Problem
Healthy cells continuously decide whether proteins should be folded, refolded, trafficked, disassembled, or destroyed. That decision process depends on chaperones, unfolded-protein responses, the ubiquitin-proteasome system, lysosomal-autophagic clearance, and enough metabolic slack to run all of it. Age reduces that slack. Stress rises while cleanup quality falls.
This is why proteostasis belongs near the center of the aging map. Proteins are not a niche molecular category. They are the machinery of signaling, repair, transport, immunity, contraction, and structural maintenance. Once protein-quality control weakens, other hallmarks become harder to contain. Mitochondrial stress worsens folding stress. Inflammation changes protein turnover pressure. Senescence alters secretory load. The failure is distributed.
Why The Aging Proteome Becomes Unstable
The proteome becomes unstable when damaged proteins arrive faster than the quality-control system can process them. Oxidative injury, glycation, translational noise, chronic inflammatory tone, and organelle dysfunction all add to the burden. At the same time, stress responses become less adaptive. Chaperone induction weakens. Proteasomal throughput can decline. Lysosomal and autophagic performance becomes more variable. The queue lengthens while the processing system slows.
That queue logic matters more than any one molecular slogan. Cells can survive significant protein damage if turnover capacity remains high enough. They fail when the maintenance ratio flips and damaged cargo starts occupying functional space faster than it can be removed. That is one reason aging often looks gradual before it looks catastrophic. Capacity erodes for years before the visible phenotype is obvious.
Autophagy Is Not The Whole Story
Recent aging discussions often elevate autophagy as if it were the full answer to proteostasis. Autophagy is important, and the 2023 hallmarks revision was right to separate disabled macroautophagy from the broader proteostasis bucket. Still, that separation does not make the larger proteostasis category disappear. Chaperone biology, proteasomal selectivity, secretory-pathway stress, endoplasmic-reticulum quality control, and extracellular protein handling still matter on their own terms.
The practical implication is that an intervention can improve one cleanup pathway without restoring the maintenance system as a whole. That is why strong claims around spermidine, fasting, heat shock, or any single proteostasis-linked tactic need to be framed narrowly. A real signal in one lane does not prove restoration of the aging proteome.
| Proteostasis layer | Main function | Common aging pressure |
|---|---|---|
| Chaperone response | Stabilize folding, refold stress-damaged proteins, triage repairable cargo | Lower inducibility and weaker response to repeated proteotoxic stress |
| Proteasome system | Tag and degrade short-lived or damaged proteins | Reduced throughput, altered selectivity, rising damaged-substrate load |
| Macroautophagy and lysosomes | Clear aggregates, organelles, and larger damaged structures | Defective cargo flux, lysosomal decline, incomplete clearance |
| Secretory and ER quality control | Manage membrane and exported proteins under high folding demand | Chronic unfolded-protein stress and weaker adaptation margins |
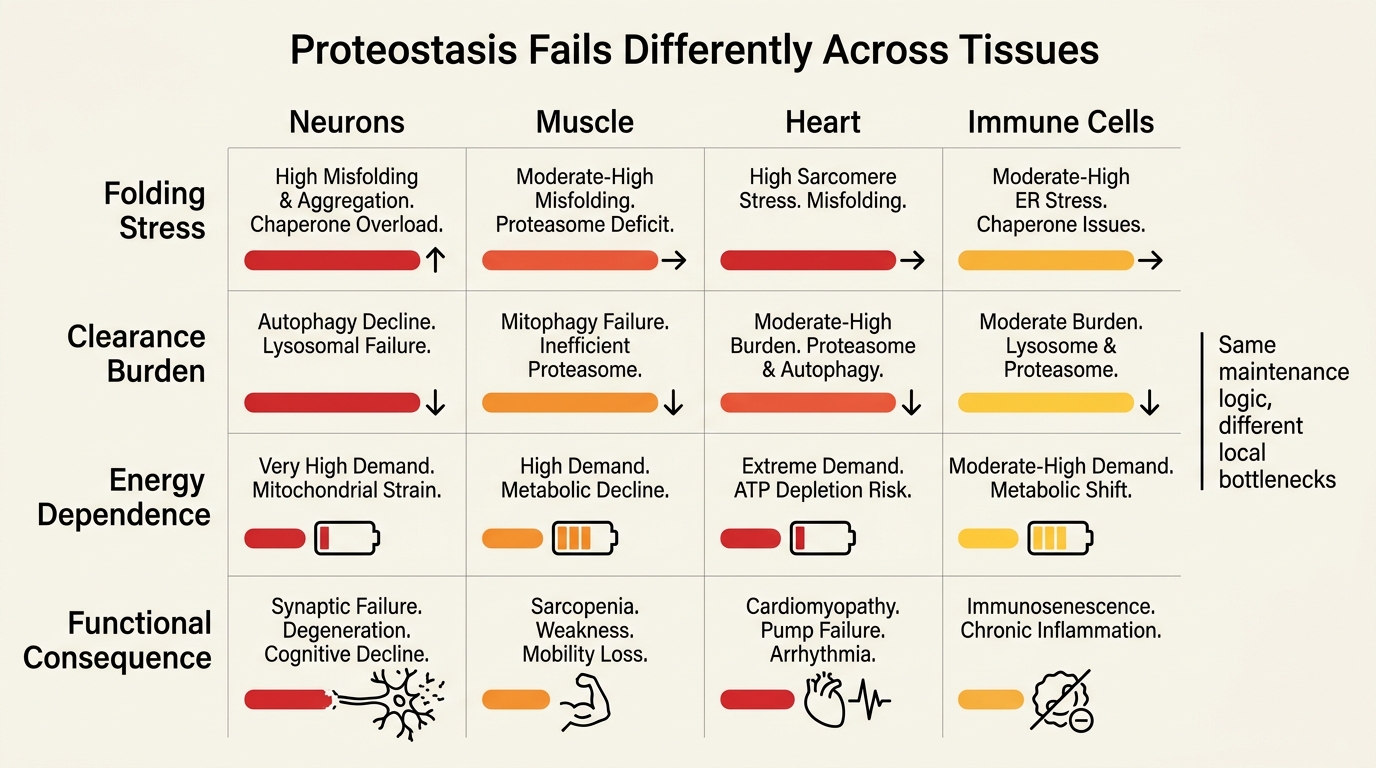
Proteostasis Failure Is Tissue-Specific In Practice
The same proteostasis theory does not look identical in every organ. Neurons are vulnerable because they are long-lived, highly specialized, and less replaceable. Skeletal muscle must maintain a large contractile protein inventory under constant turnover pressure. The heart faces relentless energetic demand with little tolerance for persistent misfolded-protein burden. Secretory tissues face a different challenge: high output with narrow folding margins. This is why proteostasis collapse does not produce one clean aging phenotype.
That tissue specificity also explains why aggregation diseases, frailty, sarcopenia, cardiometabolic decline, and neurodegeneration can share overlapping maintenance logic without being the same disease. Proteostasis is a shared vulnerability class, not a universal explanation that erases local biology.
Why Drug Translation Remains Hard
Proteostasis is attractive because it sits upstream of many visible pathologies. That same centrality creates a translation problem. A therapy that broadly increases protein turnover can easily move from helpful cleanup into unwanted stress, nutrient drain, muscle compromise, or altered signaling. A therapy that sharpens one cleanup lane may help only in a narrow tissue context. The target is real. The operating window is usually narrower than the narrative suggests.
This is why readers should place proteostasis beside the hallmarks map, mitochondrial decline, and metabolic intervention logic. Proteostasis collapse interacts with all three, but it does not collapse them into one question. Repair quality, energy state, stress signaling, and tissue reserve still have to be judged together.
What The Strongest Near-Term Use Case Looks Like
The strongest near-term use case is not a single universal anti-aging proteostasis drug. It is a more constrained strategy: identify tissues or syndromes where quality-control failure is clearly limiting, target the relevant cleanup lane, and judge success by function rather than by molecular romance. That is less cinematic than a global reset story, but it is closer to how serious maintenance biology is likely to move.
- Treat proteostasis as a systems-maintenance layer, not as a synonym for aggregates.
- Separate autophagy claims from broader protein-quality claims.
- Expect tissue-specific wins before whole-body rejuvenation claims.
- Judge interventions by preserved function, not only by proteomic or biomarker movement.
Known, Inferred, And Unknown
| Category | Assessment |
|---|---|
| Known | Loss of proteostasis is an established aging hallmark, and age is associated with weaker protein-quality control across chaperone, proteasome, and autophagic systems. |
| Known | Proteostasis failure contributes to major late-life pathologies, especially in long-lived or high-demand tissues such as brain, muscle, and heart. |
| Known | The modern field distinguishes broad proteostasis decline from disabled macroautophagy because the mechanisms overlap but are not interchangeable. |
| Inferred | Many aging interventions will disappoint if they improve one cleanup pathway but leave the wider maintenance ratio unfavorable. |
| Unknown | Which human interventions can expand proteostasis capacity meaningfully without unacceptable tradeoffs in energy balance, muscle, signaling quality, or tissue specificity. |
Source Frame
This analysis draws on stable field structure rather than one fresh headline: the 2013 and 2023 hallmarks frameworks, long-running proteostasis reviews, and the broader literature on chaperones, proteasomal degradation, lysosomal-autophagic flux, and protein-aggregation disease.
The open question is not whether proteostasis matters. It is which human interventions can improve maintenance capacity at useful scale, in which tissues, and with what durability on real functional endpoints.
Translate this longevity claim into a capital-runway decision.
Life extension logic only matters if the balance sheet can carry it. Move into WealthMeter to compare assets, spending, and yield assumptions against the same long-horizon planning problem.